Catad_tema Педиатрия - статьи
Е.В. Тозлиян, педиатр-эндокринолог, генетик, к. м. н., И.В. Шулякова, невролог, к. м. н.,
обособленное структурное подразделение «Научно-исследовательский клинический институт педиатрии» ГБОУ ВПО «Российский национальный исследовательский медицинский университет имени Н.И. Пирогова» Минздрава РФ, г. Москва
Ключевые слова:
дети, псевдогипопаратиреоз, наследственная остеодистрофия Олбрайта, ожирение, гипокальциемия, диагностика, резистентность к паратиреоидному гормону.
Keywords:
children, pseudohypoparathyroidism, Albright hereditary osteodystrophy, obesity, hypocalcemia, diagnostics, parathyroid hormone resistance.
Псевдогипопаратиреоз (греч. pseudes – ложный + гипопаратиреоз; синоним: наследственная остеодистрофия Олбрайта, синдром «яванской курицы») – редкое наследственное заболевание костной системы, имитирующее гипопаратиреоз и характеризующееся нарушением обмена кальция и фосфора; часто сопровождается задержкой умственного и физического развития. Заболевание описано впервые американским врачом-эндокринологом Albright F. в 1942 году . Распространенность заболевания составляет 7,9 на 1 млн человек .
ГЕНЕТИЧЕСКИЕ ДАННЫЕ
Псевдогипопаратиреоз (ПГП) – генетически гетерогенное заболевание. Данные о типе наследственной передачи противоречивы: как X-сцепленный доминантный , так и аутосомно-доминантный, аутосомно-рецессивный типы . В большинстве случаев развитие наследственной остеодистрофии Олбрайта связано с мутациями в расположенном на хромосоме 20 локусе 20q13 гена GNAS1 (Patten et al., 1990), кодирующего белок Gs-альфа, связанного с рецептором паратиреоидного гормона (ПТГ) . Подобный фенотип выявлен и у больных с интерстициальной делецией длинного плеча хромосомы 2 локуса 2q37 .
ПАТОГЕНЕЗ
В основе патогенеза псевдогипопаратиреоза лежит генетически обусловленная резистентность почек и скелета к действию парат-гормона в результате дефекта комплекса «специфический циторецептор – паратгормон – аденилатциклаза», что нарушает процесс образования в почках циклического 3"-, 5"-аденозинмонофосфата (цАМФ), являющегося внутриклеточным посредником действия паратгормона на метаболические процессы. Псевдогипопаратиреоз является генетически гетерогенным заболеванием. У части больных дефектен сам циторецептор, связывающий паратгормон (тип 1А псевдогипопаратиреоза), у других отмечается дефект нуклеотидсвязывающего белка, локализованного в липидном бислое клеточной мембраны и функционально связывающего рецептор с аденилатциклазой (тип 1B псевдогипопаратиреоза). У некоторых больных наблюдается ферментативная недостаточность самой аденилатциклазы (псевдогипопаратиреоз 2-го типа). Дефицит цАМФ, получающийся вследствие этих дефектов, ведет к нарушению синтеза специфических белков, определяющих биологический эффект паратгормона. Таким образом, теряется чувствительность органов-мишеней к паратгормону .
КЛИНИЧЕСКАЯ ХАРАКТЕРИСТИКА
В настоящее время выделяют 4 клинические формы патологии: типы 1А, 1В, 1С и 2. Знание их клинико-биохимических особенностей и данных генетических исследований позволяет провести дифференциальную диагностику в рамках самой нозологической формы.
Общими признаками, позволяющими заподозрить заболевание, являются диспропорциональность физического развития, низкий рост (до карликовости) за счет укорочения нижних конечностей (фото 1), брахидактилия (фото 2), круглое «лунообразное» лицо (фото 3). Иногда наблюдаются экзостозы и аплазия зубов.
Фото 1.
Внешний вид ребенка с остеодистрофией Олбрайта
(особенности фенотипа, низкий рост за счет укорочения нижних конечностей)

Фото 2.
Особенности костной системы у больного
с остеодистрофией Олбрайта
(брахидактилия – укорочение пальцев)
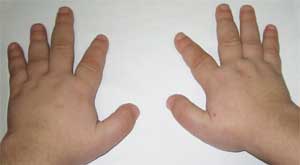
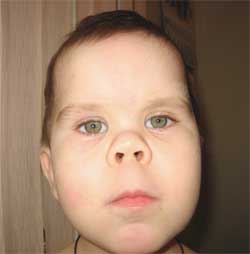
Фото 3.
Особенности фенотипа ребенка
с остеодистрофией Олбрайта
(круглое «лунообразное» лицо)
Патогномоничным признаком считается резкое укорочение I, III и V пястных и плюсневых костей (особенно III и IV), вследствие чего II пальцы на кистях и стопах оказываются длиннее остальных, а при сжатии кисти в кулак отсутствуют выпуклости в области IV и V пястно-фаланговых суставов – так называемый брахиметафалангизм. Выявляются также короткие широкие фаланги, утолщение свода черепа и деминерализация костей (остеопороз), ожирение .
Умственная отсталость (чаще умеренной степени выраженности) обнаруживается примерно у 20% больных. По данным некоторых авторов , олигофрения встречается в 70% случаев при наличии гипокальциемии и в 30% случае при нормокальциемии. Психические процессы у больных замедлены. В неврологическом статусе нередко отмечаются моторная неловкость, невротические реакции: страхи, тревога, беспокойство, плохой сон, повышение рефлексов, судороги, носящие тетанический характер и обусловленные гипокальциемией, иногда судорожные пароксизмы. Описаны также миопатические симптомы: мышечная утомляемость, мышечная слабость. Часто наблюдаются экстрапирамидные нарушения: хореиформные гиперкинезы, атетоз, лицевой гемиспазм, паркинсонизм, в отдельных случаях имеют место эпилептические пароксизмы, мозжечковые симптомы: атаксия, нарушение координации.
Нередко определяются кальцификация мягких тканей, подкожные кальцификаты (грудь, живот, пяточные сухожилия), при гистологическом исследовании которых – osteoma cutis (Izraeli et al., 1992), мозге (базальные ганглии). Важно отметить, что кальцификаты могут быть уже при рождении. Вследствие гипокальциемии обычно развивается катаракта и возникают дефекты эмали зубов.
ПСЕВДОГИПОПАРАТИРЕОЗ ТИПА 1А
имеет аутосомно-доминантный тип наследования. Ген псевдогипопаратиреоза типа 1А – GNAS1 – локализован на длинном плече хромосомы 20, в локусе 20q13.2. Развитие заболевания связано с дефицитом гуанин-нуклеотид-связывающего белка (Gs-белок). При этом ПТГ, связываясь с рецепторами тканей-мишеней, не способен активизировать циклический аденозинмонофосфат (цАМФ) и вызвать тканевой ответ. Вероятно, подобный механизм лежит в основе развития нечувствительности тканей других органов и эндокринных желез (гипофункция щитовидной железы, гонад, гипофиза, сахарный диабет, а также сниженный ответ печени на введение глюкагона), наблюдаемой при псевдогипопаратиреозе типа 1А. При данном типе патологии не наблюдается характерной для нормы повышенной экскреции цАМФ с мочой в ответ на экзогенное введение ПТГ. Заболевание диагностируется чаще в возрасте 5–10 лет. У больных наблюдаются низкий рост, короткая шея, круглое лицо, укорочение метакарпальных и метатарзальных костей (чаще укорочение IV и реже II пальцев) – так называемый брахи-метафалангизм. Отмечаются кальцификация мягких тканей, подкожные кальцификаты, которые могут выявляться уже при рождении; нередко наблюдается одновременное вовлечение других эндокринных желез: щитовидной железы (гипофункция), гонад, поджелудочной железы (сахарный диабет). Вследствие гипокальциемии нередко развиваются катаракта и дефект эмали зубов. В качестве дифференциально-диагностического теста отличия ПГП типа 1А от гипопаратиреоза: отсутствие клинического эффекта от парентерального введения ПТГ в виде подъема уровня кальция в крови и увеличения почечной экскреции фосфора с мочой (фосфатурический эффект).
При биохимическом исследовании выявляются гипокальциемия, гиперфосфатемия, увеличение уровня паратиреоидного гормона в крови, гипофосфатурия. Уровень Gs-белка в крови снижен. При рентгенологическом исследовании костной системы обнаруживаются укорочение метакарпальных и метатарзальных костей, генерализованная деминерализация, утолщение костей свода черепа.
ПСЕВДОГИПОПАРАТИРЕОЗ ТИПА 1B
имеет аутосомно-доминантный тип наследования, однако не исключен доминантный, сцепленный с Х-хромосомой тип наследования. Необходимо иметь в виду наблюдающуюся иногда неполную пенетрантность гена болезни и возможность скрытого носительства патологии. Поэтому рекомендуется клиническое (выявление субклинического течения болезни) и биохимическое обследование (определение уровней кальция, фосфора, ПТГ крови) предполагаемых носителей заболевания. ПГП типа 1В обусловлен дефицитом тканевых рецепторов к паратиреоидному гормону в органах-мишенях и ограниченной резистентностью к паратгормону. Клиническая картина сходна с клиникой типа 1А, но отсутствует поражение других эндокринных желез, реже встречается остеодистрофия.
У больных отсутствует реакция почек на экзогенное введение паратиреоидного гормона в виде увеличения экскреции циклического аденозинмонофосфата с мочой, однако, в отличие от типа 1А, уровень Gs-белка в крови нормален. Женщины поражаются чаще мужчин, однако тяжесть заболевания может быть одинаковой как у мужчин, так и у женщин.
ПСЕВДОГИПОПАРАТИРЕОЗ ТИПА 1С
некоторые авторы отождествляют с псевдо-псевдогипопаратиреозом (ППГП), описанным Albright F. в 1952 году. Характеризуется свойственной ПГП клинической картиной, однако уровни кальция, фосфора в крови и моче остаются в пределах нормы. Показатели ПТГ и Gs-белка в крови также сохраняются на нормальном уровне. У некоторых больных с ПГП типа 1С обнаруживаются делеции de novo на хромосоме 2 . Не исключено, что этот вариант болезни является подтипом ПГП типа 1А.
ПСЕВДОГИПОПАРАТИРЕОЗ ТИПА 2
клинически сходен с другими типами заболевания, однако имеет аутосомно-рецессивный тип наследования. Не исключено существование и аутосомно-доминантных форм патологии. Патогенез развития связан с внутриклеточной резистентностью к цАМФ. ПТГ при этом связывается с рецепторами и вызывает нормальную ответную реакцию клеток на ПТГ в виде увеличения экскреции цАМФ. Внутриклеточная нечувствительность к цАМФ, однако, не позволяет осуществиться полной реализации действия ПТГ. При этом сохраняется нормальная реакция почек на экзогенное введение паратиреоидного гормона в виде увеличения экскреции циклического аденозинмонофосфата с мочой. Высказывается мнение, что ПГП типа 2 может быть связан с дефицитом витамина D .
Таким образом, выделенные типы ПГП клинически характеризуются пониженной чувствительностью органов-мишеней к ПТГ, однако различаются патогенетическими механизмами формирования нечувствительности тканей.
ДИАГНОСТИКА
Лабораторным дифференциально-диагностическим тестом может служить характер почечной экскреции цАМФ в ответ на введение ПТГ: повышенная экскреция цАМФ отмечается при типе 2 и ее отсутствие – при типе 1. Диагноз подтверждается обнаружением сниженного уровня гуанин-нуклеотидсвязывающего белка (Gs-белок) в крови (в среднем в 1,5–2 раза) по сравнению с нормой. Гипокальциемия, как правило, сочетается с гиперфосфатемией и гипофосфатурией. Уровень ПТГ повышен; при 1C-типе уровень ПТГ в норме, что дало основание для названия «псевдогипопаратиреоз». При рентгенологическом исследовании костной системы обнаруживаются укорочение пястных и плюсневых костей, нередко генерализованная деминерализация (остеопороз), утолщение костей свода черепа. В дерматоглифическом рисунке отмечается смещение осевого ладонного трирадиуса.
Критерии диагноза:
ЛЕЧЕНИЕ И ПРОФИЛАКТИКА
Лечение при гипокальциемии заключается в назначении препаратов кальция в дозах, достаточных для поддержания нормальной концентрации кальция в крови. Большое значение имеет терапия витамином D. В настоящее время применяют активные метаболиты витамина D – оксидевит, 1-альфа-Д3, кальцитрин и др. в дозе 1–2 мкг/сутки с положительным результатом (увеличение содержание кальция в крови, уменьшение проявлений судорожного синдрома). Эффективен также тахистин (0,5–1,5 мг/сутки). Данный препарат увеличивает всасывание кальция в кишечнике и тем самым способствует повышению уровня кальция в крови. Противосудорожная терапия используется как дополнительное лечение. На интеллектуальное развитие лечение не оказывает заметного действия, но наряду с уменьшением симптомов судорожного синдрома наблюдается регресс неврологических проявлений (подкорковых нарушений, хореиформных гиперкинезов, атетоза и др.). Во избежание передозировки препаратов витамина D необходим контроль концентрации кальция в крови каждые 3–7 дней в течение первых 2 недель лечения и каждый месяц в течение последующих 2–3 месяцев. По достижении стабильной концентрации кальция в крови достаточно проверять ее 1 раз в 2–3 месяца. Диета с ограничением фосфора помогает нормализовать как концентрацию фосфора, так и содержание кальция в крови и устранить симптомы вторичного гиперпаратиреоза. При недостаточности других желез внутренней секреции проводят заместительную терапию соответствующими гормонами.
Лечение паратгормоном неэффективно. Для купирования судорожных приступов внутривенно вводят 10%-ный раствор кальция хлорида или кальция глюконата; внутрь – 5–10%-ный раствор кальция хлорида по 1 столовой ложке 3–4 раза в день: кальция глюконат, кальция лактат – до 10 г в день.
ПРОГНОЗ для жизни определяется выраженностью судорожного синдрома.
ПРОФИЛАКТИКА болезни основывается на данных медико-генетического консультирования.
МЕДИКО-ГЕНЕТИЧЕСКОЕ КОНСУЛЬТИРОВАНИЕ
При медико-генетическом консультировании следует исходить из аутосомно-доминантного типа наследования и высокого (50%) риска повторения заболевания в семье при унаследованных формах. С целью идентификации характера типа наследования необходимо проводить тщательное обследование родителей, так как синдром может проявляться минимальными клиническими симптомами. В настоящее время разработана и совершенствуется молекулярно-генетическая диагностика заболевания путем типирования мутаций в гене GNAS1 на хромосоме 20. Разрабатываются способы пренатальной диагностики заболевания в целом и отдельных его типов.
КЛИНИЧЕСКОЕ НАБЛЮДЕНИЕ Мальчик Г., 14,5 лет (фото 4), поступил в Научно-исследовательский клинический институт педиатрии с диагнозом: дегенеративное заболевание нервной системы? врожденная наружная гидроцефалия; симптоматическая эпилепсия; наследственный синдром? болезнь накопления? метаболическая энцефалопатия; субклинический гипотиреоз; низкорослость смешанного генеза; когнитивные нарушения.
Жалобы при поступлении на интенсивные приступообразные головные боли, локализующиеся в лобной области и сопровождающиеся рвотой, которая приносит облегчение, снижение памяти и успеваемости в школе, судорожные приступы, во время которых происходит подергивания в правой руке.
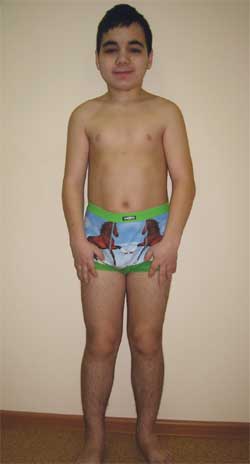
Фото 4.
Ребенок Г., 14,5 лет, с остеодистрофией Олбрайта
(особенности фенотипа, низкий рост, укорочение конечностей, брахидактилия)
Анамнез семейный: родители – армяне по национальности, не состоящие в кровном родстве и не имеющие профессиональных вредностей. В родословной случаев психических заболеваний, эпилепсии, задержки в развитии – не отмечалось. Сибс, сестра 17 лет – со слов – здорова.
Анамнез жизни и заболевания:
мальчик от 2-й беременности, протекавшей без особенностей, роды вторые, в срок, физиологические, масса при рождении – 3100 г, длина – 51 см. Закричал сразу, оценка по шкале Апгар – 7/9 баллов. Ухудшение состояния на 3-и сутки – судороги неонатальные, купированы в роддоме. Ранний постнатальный период – без особенностей. Отмечалась незначительная темповая задержка моторного развития на первом году жизни, самостоятельная ходьба с 1 года 3 мес. В связи с чем наблюдался неврологом с диагнозом: органическое поражение ЦНС; врожденная гидроцефалия; неонатальные судороги; фебрильные судороги в анамнезе.
Получал диакарб, финлепсин. Дебют приступов с 1 года 11 мес. – асимметричные, тонические в виде напряжения правой руки и ноги, с заведением глаз, до 2 мин., без потери сознания, частые до 10 эпизодов в сутки. Получал депакин нерегулярно. На фоне самостоятельной отмены – однократный тонический статус. В 2 года проведена по месту жительства КТ головного мозга, где выявлены единичные очаги демиелинизации в затылочных долях.
Консультирован нейрохирургом, рекомендовано консервативное лечение. С 3 лет отмечается задержка психоречевого развития, рекомендовано наблюдение психиатра.
С 4–5 лет родители стали отмечать деформацию и укорочение пальцев стоп и кистей, в особенности II–IV пальцев симметрично на руках и ногах, снижение ростовых показателей. В 8 лет заключение логопеда – общее нарушение речи 2–3-го уровня, рекомендовано обучение в специализированной школе. В этом же возрасте осмотр генетиком по месту жительства, заключение: наследственная болезнь обмена? рекомендовано исследование аминокислот крови, изменений не было выявлено; окончательное заключение: данных за наследственное заболевание обмена не выявлено; гипохондроплазия; рекомендовано лечение невролога и эндокринолога.
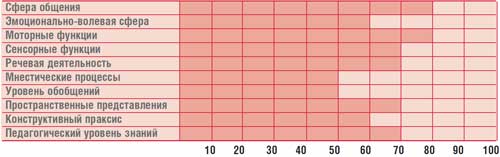
Таблица.
Профиль психического развития ребенка Г., 14,5 лет (IQ = 68)
В возрасте 8 лет консультирован эндокринологом по поводу задержки роста и развития. При рентгенологическом исследовании кистей рук отмечены особенности: средние, основные фаланги и пястные кости укорочены, утолщены; диагноз рентгенолога – ахондроплазия.
Неоднократно обследуется по месту жительства в неврологическом стационаре. В 12 лет появились судорожные приступы без потери сознания с подергиванием правой руки, носящие серийный характер, назначена противосудорожная терапия (депакин), частота приступов значительно сократилась. В 13 лет проведена МРТ головного мозга с контрастированием – симметричные изменения в основании височных долей на уровне ядер в виде повышения МР-сигнала, что характерно для токсических (марганец) или метаболических (медь, железо) энцефалопатий.
Вновь осмотрен в возрасте 13 лет 3 мес. эндокринологом, при исследовании тиреоидного профиля выявлено повышение тиреотропного гормона (ТТГ), диагностирован субклинический гипотиреоз, назначен L-тироксин.
При анализе амбулаторной карты ребенка и документации по месту жительства исследование кальция и фосфора проводилось однократно, в возрасте 1,5 лет, отмечалась гипокальциемия, но по данному поводу дообследование не проводилось. Учитывая неопределенность диагноза по месту жительства, генетиком ребенок направлен в Москву, в Научно-исследовательский клинический институт педиатрии, с целью уточнения диагноза.
Данные объективного исследования:
Рост – 143 см, масса – 43 кг.
Физическое развитие очень низкое, гармоничное, телосложение диспропорциональное за счет укорочения конечностей. Sds роста соответствует –2,8 отклонениям от нормы (норма –2+2).
Особенности фенотипа: круглое лицо, короткая шея, антимонголоидный разрез глазных щелей, широкое переносье, высокий лоб, брахидактилия, укорочение IV и V пястных и плюсневых костей (фото 5). По внутренним органам – без особенностей. Половое развитие – Tanner III–IV стадия (что соответствует возрасту).
Данные лабораторных и функциональных исследований:
Клинический анализ крови и мочи – норма.
Биохимический анализ крови: общий кальций – 1,39 (норма 2,02–2,6 ммоль/л), кальций ионизированный – 0,61 (норма 1,13–1,32 ммоль/л), фосфор неорганический – 3,66 (норма 0,86–1,56 ммоль/л), остальные показатели в пределах нормы.
Биохимический анализ мочи: почечная экскреция фосфатов снижена – 11,5 ммоль/л (норма 19–32 ммоль/л).
Тиреоидный профиль: ТТГ – 11,75 (норма 0,4–4,0 мкМЕ/мл), свободный Т4 – 0,49 (норма 1,0–1,8 нг/дл).
Паратиреоидный гормон – 499 (норма 12– 65 пг/мл), СТГ – 7 нг/мл (норма 7–10 нг/мл), соматомедин-С – 250 нг/мл (норма 88–360 нг/мл).
УЗИ внутренних органов – без особенностей.
ЭКГ – миграция суправентрикулярного водителя ритма на фоне регулярной ЧСС 71– 80 уд/мин. Неполная блокада правой ножки пучка Гиса. Нарушение процесса реполяризации в миокарде задней стенки левого желудочка (снижение з.Т III, аVF).
R-графия позвоночника – правосторонний сколиоз грудного отдела позвоночника 1-й степени, выраженный остеопороз.
R-графия кистей рук с захватом предплечий – укорочение и расширение концевых и средних фаланг. Костный возраст – 13,5– 14 лет.
ЭЭГ-паттернов эпилептической активности не зарегистрировано.
МРТ головного мозга – МР-картина множественных субкортикальных очагов повышенного МР-сигнала в лобных долях, наружная компенсированная гидроцефалия с атрофией вещества головного мозга.
МСКТ головного мозга – симметричные участки обызвествления лентиформных ядер. Диффузные гиперденсивные участки в таламусах, хвостатых ядрах с участком обызвествления справа. Множественные точечные обызвествления покровных мягких тканей черепа.
Аудиограмма – без патологии.
ДНК-диагностика в гене GNAS1 – в работе.
Консультации специалистов:
Эндокринолог – наследственная остеодис-трофия Олбрайта типа 1А (псевдогипопара-тиреоз). Первичный гипотиреоз, неполная медикаментозная компенсация.
Окулист – катаракта полная вторичная. Рекомендовано оперативное лечение.
Психолог – когнитивные нарушения (психологический профиль ребенка представлен в табл.).
Учитывая фенотип ребенка, данные анамнеза, результаты дополнительных исследований (гипокальциемия, гиперфосфатемия, гипофосфатурия, повышение паратиреоидного гормона крови), кальцинаты в веществе головного мозга, наличие катаракты, гипотиреоза), поставлен диагноз: наследственная остеодистрофия Олбрайта 1А-типа (псевдогипопаратиреоз). Рекомендовано проведение ДНК-диагностики - поиск мутаций в гене GNAS1.
Лечение: ребенку рекомендован прием эутирокса в дозе 100 мкг/сутки; активный метаболит витамина Д - альфа-Д3 («Тева») в дозе 2 мкг/сутки; кальций («Сандоз») 2000 мг/сутки; постоянный прием противосудорожной терапии - финлепсин 800 мг/сутки под наблюдением невролога-эпилептолога; занятия с логопедом-дефектологом и психологом; энерготропная терапия (Элькар и коэнзим Q10 в возрастных дозах). Контроль показателей фосфорно-кальциевого обмена, уровня паратгормона.
Таким образом,
представленное клиническое наблюдение демонстрирует сложности дифференциально-диагностического поиска, важность своевременного исследования простых биохимических параметров (при эпилепсии обязателен неоднократный скрининг показателей фосфорно-кальциевого обмена), исходы поздней диагностики генетически детерминированного заболевания, необходимость интегрировать отдельные признаки в общий фенотип того или иного патологического состояния для целенаправленной своевременной диагностики отдельных форм наследственных заболеваний. Своевременная диагностика, уточнение генеза каждого синдрома особенно важны, так как позволяют найти оптимальный подход к лечению этих состояний, профилактике возможных осложнений (вплоть до инвалидности ребенка); предупреждение повторного возникновения наследственных болезней в пораженных семьях (медико-генетическое консультирование). Это диктует необходимость врачам различных специальностей четко ориентироваться в потоке наследственно обусловленной патологии. Список литературы находится в редакции.
Синдром Олбрайта, или иначе псевдогипопаратиреоз - это очень редкое генетически детерминированное заболевание, которое проявляется нарушениями кальциевого и фосфорного обмена и, как следствие, патологией костной системы. Как правило, основные симптомы сопровождаются еще и отставанием в развитии - как умственном, так и физическом. Чаще заболевание встречается у женщин.
Ученые считают, что синдром Олбрайта обусловлен интактностью клеток почек и костей к гормонам паращитовидных желез. Это связано с дефектом рецепторов. В почках нарушается процесс синтеза цАМФ (циклического аминазинмогофосфата), который является вспомогательным звеном в метаболических процессах, связанных с паратгормоном. Такие повреждения появляются при повреждении специфических генов.
Нарушение может быть на любом участке метаболической цепи. У одних больных поврежден сам рецептор, у других изменениям подвергаются белки клеточной мембраны, у третьих заболевание связано с недостаточной выработкой цАМФ. Все эти случаи связывает генетическая обусловленность патологии.
Синдром Олбрайта тесно связан с физиологией паращитовидных желез. В норме они регулируют уровень кальция и фосфора в крови, но при данном патологическом состоянии они равнодушны к колебаниям этих элементов, что может привести к возникновению вторичного гиперпаратиреоза. Но даже высокие уровни парагормона не стимулируют ускоренное элиминирование фосфора и цАМФ из крови (так как почки интактны к его воздействию) и в тоже время провоцируют изменения в структуре костей.
Как правило, наблюдается компенсаторное увеличение паращитовидных желез, пористость костей, появление в них кистозных образований, кальцификатов в коже, жировой ткани, мышцах, сердце, сосудах и конъюнктиве и роговице.

Синдром Олбрайта проявляется практически так же, как и истинный гипопаратиреоз. У больных наблюдаются судороги, которые могу возникать как сами по себе, так и после воздействия раздражающих факторов (нервное напряжение, перепад температур, тяжелая физическая работа и т.д.). Кальцинаты, располагающиеся в коже и подкожно-жировой клетчатке, могут изъязвляться и доставлять дискомфорт пациентам. При этом больной будет обращаться за помощью не к эндокринологу или генетику, а к хирургу или дерматологу.
Такие люди, как правило, низкого роста, у них круглое одутловатое лицо, ожирение и укорочение пальцев. Наблюдаются ложные суставы там, где их быть не должно, отсутствует подвижность в тех местах, где она предполагается анатомически, нарушение конфигурации суставов и костей. Среди симптомов также рвота, кровь в моче, катаракта и изменения в зубной эмали.
Кроме того, из-за низкого уровня кальция в крови с детства у пациентов наблюдается снижение интеллекта и задержка развития.

Синдром Олбрайта - болезнь, которую довольно тяжело диагностировать у младенца, но в возрасте 5-10 лет появляются достаточно яркие проявления, которые не оставляют у врача сомнений в диагнозе. У детей наблюдаются множественные пороки развития костной системы, в крови снижен уровень кальция и повышен уровень фосфора и паратгормона.
Для подтверждения диагноза можно провести тест на количество фосфатов и цАМФ в моче. Для этого пациенту вводится 200 единиц паратгормона внутривенно и затем через 4 часа собирается порция мочи. Если достоверного повышения не наблюдается, то это может свидетельствовать о резистентности почечной ткани к паратгормону.
Этих признаков достаточно, чтобы поставить синдром Олбрайта. Диагностика может быть дополнена рентгенографией. Она дает возможность увидеть специфические изменения в костной системе и мягких тканях пациента.
Кроме паратгормона у людей с таким диагнозом регистрируют резистентность и к другим гормонам, поэтому женщинам проводят дифференциальную диагностику с синдромом Шерешевского-Тернера. Эти два заболевания имеют внешнюю схожесть, и для верификации диагноза врач должен назначить анализ на половой хроматин и дать направление на гинекологическое обследование и УЗИ органов малого таза.

После того как будет поставлен диагноз «синдром Олбрайта», лечение необходимо назначать, не откладывая, так как задержки усугубляют дефицит умственного развития. Детям назначают препараты кальция в достаточных суточных дозах, чтобы поддерживать нормальную концентрацию в сыворотке крови. Затем присоединяют к этому прием витамина Д в дозе, не превышающей 100 000 единиц в день.
Для корректировки лечения необходимо контролировать уровень кальция каждые 7 дней в течение первых двух недель, а затем раз в месяц до конца терапии. Когда оптимальная концентрация кальция в крови будет достигнута, можно проверять его раз в квартал.
Чтобы устранить симптомы вторичного гиперпаратиреоза, рекомендуется соблюдать диету со сниженным количеством фосфора. При обнаружении недостаточности других гормонов проводят заместительную и симптоматическую терапию.
Если лечение начато во время и терапия рациональная, то прогноз для жизни благоприятный. Но перед беременностью женщинам с таким диагнозом необходимо пройти консультацию у генетика.
Синдром Олбрайта – это наследственная патология с неясным происхождением. Ее еще называют псевдогипопаратиреозом. Для него характерно поражение костной ткани. Заболевание проявляется в основном у женской половины населения, хотя встречается и у мальчиков. Практически у всех больных отмечаются отклонения в умственном и физическом развитии. Кости человека начинают разрушаться под влиянием обменных процессов фосфора и кальция. Начинается выделение паратгормона паращитовидной железой.
Основная причина такой патологии, как синдром Форбса – Олбрайта – это наследственная устойчивость тканей организма к влиянию паратгормона. Если человек не болен, тогда гормон действует посредством определенного вещества. Когда заболевание проявляется, то состав этого веществ изменяется на генетическом уровне. Таким образом периферические ткани становятся нечувствительными к паратгормону.
Синдром Олбрайта имеет три основных симптома:
Заболевание псевдогипопаратиреоз проявляет симптомы специфического характера.
При заболевании псевдогипопаратиреозом проводится диагностика, основанная на визуальном осмотре.
Также применяются лабораторные и аппаратные исследования:
Больные, страдающие от синдрома Олбрайта, должны регулярно проходить осмотры у таких специалистов, как эндокринолог, окулист, гинеколог, травматолог.
Псевдогипопаратиреоз лечится комплексно.
Так как это генетическая патология, то терапия будет направлена на устранение появившихся симптомов:
Для каждого больного, страдающего от синдрома Олбрайта, способ терапии подбирается в индивидуальном порядке. Пациент должен строго соблюдать все рекомендации и назначения врача.
Проводится периодический контроль уровня кальция в организме пациента. На начальном этапе лечения такое исследование проводится один раз в неделю. Постепенно частота замеров снижается до одного раза в месяц. Она будет проводиться до конца назначенного лечения.
Если диагностирован псевдогипопаратиреоз, тогда больному необходимо придерживаться строгой диеты. Она заключается в полном исключении из меню продуктов, в которых присутствует фосфор.
Профилактика заключается в следующем:
Такая патология диагностируется у одного человека на один миллион жителей планеты. К сожалению, на сегодняшний день еще нет специфической терапии. Существует лишь симптоматическое лечение. Но если пациент будет находиться под контролем врача, есть шанс на благоприятный исход.
Наследственная остеодистрофия Олбрайта (синонимы: псевдогипопаратиреоз типа 1а, болезнь Олбрайта, синдром Олбрайта) относится к редким заболеваниям костной системы, обусловленным мутацией гена, расположенного в 20 хромосоме. Патология характеризуется нарушением кальциевого и фосфорного обмена, что сказывается задержкой физического и иногда умственного развития. О типе наследования синдрома Олбрайта еще ведется дискуссия.
Для сведения: первым описал заболевание американский эндокринолог F. Albright в 1942 году. В настоящее время частота патологии составляет 79 случаев на 10 миллионов человек. Женщины болеют в два раза чаще, чем мужчины.
Заболевание имитирует гипопаратиреоз - недостаточность гормона паращитовидной железы (паратгормона). Однако механизм возникновения отличается тем, что дефицита гормона нет, но генетический дефект комплекса клеточных рецепторов в тканях почек и скелета блокирует его действие. Метаболические процессы, которые регулирует в норме паратгормон через синтез цАМФ, становятся невозможными. Паращитовидные железы при этом могут быть компенсаторно увеличены.

В зависимости от типа заболевания (а сейчас выделяют четыре клинические формы: 1 и 2 тип, первый еще подразделяют на подтипы А, В и С), возможны различные варианты блокировки действия паратгормона, а также нарушение резистентности к другим гормонам: глюкагону, ТТГ, гонадолиберину, АДГ. В результате развиваются явления гипотиреоза, аменорея, нарушается концентрационная функция почек.
В крови повышается количество фосфора, а уровень активной формы витамина D3 и кальция снижается.
Заболевание приводит ко множественным аномалиям развития скелета, которые обнаруживаются в детском возрасте, иногда после года, но обычно в 5-10 лет и даже позже. Заподозрить болезнь Олбрайт у ребенка можно по таким признакам:

Важно! Любой из этих симптомов требует обращения к эндокринологу, назначения диагностических мероприятий для выявления причин состояния и подбора адекватной терапии. С 90-х годов прошлого века диагностика проводится на основе методов молекулярной генетики.
Несвоевременное обращение к эндокринологу и генетику задерживает постановку правильного диагноза и возможность лечения.
В костной ткани обнаруживаются изменения, сходные с проявлениями гипопаратиреоза:
Кальций высвобождается из костей и формирует кальцинаты в мягких тканях:
На коже выделяются участки гиперпигментации.
Нарушение обмена кальция приводит к появлению судорог и рвоты. В ряде случаев наблюдается гематурия из-за формирования оксалатных кристаллов в мочевыводящих путях.

Задержка умственного развития, сахарный диабет, катаракта также могут быть проявлениями болезни Олбрайта.
Постановка диагноза основана на анализе клинических проявлений, рентгеновском обследовании костей и мягких тканей, выявлении псевдогипопаратиреоза с помощью биохимических тестов.
Анализ крови показывает гипокальциемию и гиперфосфатемию, уровень активности щелочной фосфатазы сыворотки в норме или повышен, как уровень ПТГ.
Лабораторным доказательством псевдогипопаратиреоза является отсутствие повышения в моче уровня нефрогенного цАМФ и фосфатов в ответ на введение паратгормона.
В отличие от истинного гипопаратиреоза при болезни Олбрайта введение паратгормона не дает эффекта.
Целью лечения является восстановление уровня кальция и витамина D в крови до нормы. Для этого назначают прием препаратов, содержащих эти вещества: дигидротахистерол, оксидевит, кальцитрин.
Синдром Олбрайта – редкое генетическое заболевание с невыявленным происхождением. Передается по наследству и в основном характеризуется поражением костной системы. Чаще всего встречается у представительниц женского пола. Но бывает, когда заболеть могут и мальчики. Большинство пациентов с таким синдромом страдают физической и умственной отсталостью.
Название заболевание получило по фамилии ученого, который его открыл. Костная система начинает разрушаться под воздействием метаболизма кальция и фосфора. Паращитовидная железа выделяет паратгормон, у человека с синдромом Олбрайт резистентность тканей к паратгормону нарушена.
Выделяют три основных признака заболевания. При наличии даже двух из трех можно ставить такой диагноз.

 Пациенты, страдающие синдромом Мак-Кьюна
, должны все время находиться под наблюдением узких специалистов (гинеколог, эндокринолог, травматолог, окулист).
Пациенты, страдающие синдромом Мак-Кьюна
, должны все время находиться под наблюдением узких специалистов (гинеколог, эндокринолог, травматолог, окулист).
Необходимо комплексное лечение. Так как болезнь характеризуется генетической мутацией, то необходимо в первую очередь избавляться от симптомов его проявления.
Показано обязательное гормональное лечение для поддержания эндокринной системы.
Необходимо не упустить момент, когда начнут деформироваться кости лица, чтобы предотвратить эти изменения во время болезни.
Иногда требуется и хирургическое вмешательство . Его назначают в том случае, когда из-за изменений в строении черепа возникает риск снижения слуха или зрения.
Во время болей используют специальные обезболивающие средства, содержащие небольшое количество бисфосфонатов.
Очень важно проводить профилактические меры по укреплению мускулатуры.
Для каждого пациента, страдающего болезнью Олбрайта, врач подбирает свое индивидуальное лечение, которое требуется соблюдать неукоснительно.
Необходимо постоянно контролировать количество кальция в организме. При начале проведения терапии такое обследование проводится раз в неделю, затем их частота сокращается до одного раза в месяц, пока не закончится основное лечение.
Очень важно во время лечения соблюдать диету, при которой количество продуктов, содержащих фосфор, урезается до минимума.
Следует заметить, что, женщина, страдающая этим синдромом , при решении забеременеть обязательно должна проконсультироваться с хорошим генетиком.
Болезнь Олбрайта (Мак-Кьюна) встречается у одного человека на 1 000 000. Специфического лечения для этого синдрома еще не изобрели, но не стоит терять надежды. Медицина не стоит на месте, а врачи помогут преодолеть неприятные моменты и будут держать пациента все время под наблюдением.








