Миелоидный лейкоз или миелолейкоз – это опасное онкозаболевание системы кроветворения, при котором поражаются стволовые клетки костного мозга. В народе лейкоз часто именуют «белокровие». Как следствие, они полностью прекращают выполнять свои функции и начинают стремительно умножаться.
В костном мозге человека продуцируются , и . Если пациенту ставят диагноз – миелолейкоз, то в крови начинают созревать и быстро размножаться патологически изменённые незрелые клетки, которые в медицине именуют бласты. Они полностью блокируют рост нормальных и здоровых кровяных клеток. Через некоторый промежуток времени рост костного мозга полностью прекращается и данные патологические клетки посредством кровеносных сосудов попадают ко всем органам.
В начальной стадии развития миелолейкоза происходит значительное увеличение количества зрелых лейкоцитов в крови (до 20 000 в мкг). Постепенно их уровень увеличивается в два и больше раз, и достигает 400 000 в мкг. Также при данном недуге происходит увеличение в крови уровня , что свидетельствует о тяжёлом течении миелоидного лейкоза.
Этиология острого и хронического миелолейкоза на сегодняшний день ещё до конца не изучена. Но учёные со всего мира работают над решением этой проблемы, чтобы в дальнейшем была возможность предотвратить развитие патологии.
Возможные причины развития острого и хронического миелолейкоза:
Этиология развития острого и хронического миелолейкоза продолжает изучаться по сей день.
Миелоидный лейкоз в медицине делят на две разновидности:
Острый миелоидный лейкоз – заболевание крови, при котором происходит неконтролируемое размножение лейкоцитов. Полноценные клетки заменяются на лейкемические. Патология быстротекущая и без адекватного лечения человек может умереть через несколько месяцев. Продолжительность жизни пациента напрямую зависит от стадии, на которой будет обнаружено наличие патологического процесса. Поэтому важно при наличии первых симптомов миелоидного лейкоза обратиться к квалифицированному специалисту, который проведёт диагностику (наиболее информативным является анализ крови), подтвердит или опровергнет диагноз. Острым миелоидным лейкозом болеют люди из разных возрастных групп, но наиболее часто она поражает лиц старше 40 лет.
Симптомы заболевания, как правило, проявляются практически сразу. В очень редких клинических ситуациях состояние больного ухудшается постепенно.
При наличии одного или нескольких таких симптомов рекомендовано как можно скорее посетить медицинское учреждение. Важно помнить, что прогноз заболевания, а также продолжительность жизни пациента, у которого его выявили, во многом зависит именно от своевременно проведённой диагностики и лечения.
Хронический миелоидный лейкоз – это злокачественный недуг, поражающий исключительно гемопоэтические стволовые клетки. Генные мутации происходят в незрелых миелоидных клетках, которые, в свою очередь, продуцируют эритроциты, тромбоциты и практически все виды белых клеток крови. Как следствие, в организме образуется аномальный ген, именуемый BCR-ABL, являющийся чрезвычайно опасным. Он «атакует» здоровые кровяные клетки и преобразует их в лейкозные. Место их локализации – костный мозг. Оттуда с током крови они распространяются по всему организму и поражают жизненно важные органы. Хронический миелолейкоз развивается не стремительно, для него характерно длительное и размеренное течение. Но главная опасность состоит в том, что без должного лечения может перерасти в острый миелоидный лейкоз, который за несколько месяцев способен убить человека.
Болезнь в большинстве клинических ситуаций поражает людей из различных возрастных групп. У детей же возникает эпизодически (случаи заболеваемости очень редки).
Хронический миелоидный лейкоз протекает в несколько стадий:

Дополнительные методики:
При выборе определённого метода лечения для данной болезни, необходимо учитывать стадию её развития. Если заболевание выявлено на раннем этапе, то пациенту обычно назначают общеукрепляющие препараты и сбалансированное питание, богатое витаминами.
Основной и наиболее эффективный способ лечения – медикаментозная терапия. Для лечения используют цитостатики, действие которых направлено на остановку роста опухолевых клеток. Также активно используется лучевая терапия, пересадка костного мозга и переливание крови.
Большинство способов лечения данной болезни вызывают довольно тяжёлые побочные эффекты:
Для лечения заболевания и продления жизни пациента используют следующие химиотерапевтические препараты:
Выбор медикаментов напрямую зависит от стадии недуга, а также от индивидуальных особенностей пациента. Все препараты назначаются строго лечащим врачом! Самостоятельно корректировать дозу строго запрещено!
К полному выздоровлению может привести только трансплантация костного мозга. Но в таком случае стволовые клетки пациента и донора должны быть на 100% идентичны.
Развитие опухолевых процессов с каждым годом набирает обороты. Большинство ученых мира изучают возможные факторы развития состояния и основные методы терапии для излечения пациентов, а профилактическими мероприятиями занимаются все виды лечебных учреждений. Развитие может коснуться любого органа или системы организма. Миелолейкоз - что это такое? Основные причины этого заболевания, методы диагностики и терапии рассмотрим далее.
Основывается на созревании молодых клеток - тромбоцитов, эритроцитов и лейкоцитов в Параллельно с этим процессом происходит уничтожение старых клеток печенью и селезенкой.
Форменных элементов в крови столько же, сколько и плазмы. При этом самое большое количество приходится на белые кровяные клетки - лейкоциты. Они отвечают за реакцию организма на воздействие чужеродных агентов и соединений и позволяют поддерживать иммунную систему на должном уровне.
Неконтролируемая выработка большого количества лейкоцитов называется миелоидным лейкозом. Это опухолевое заболевание, которое сопровождается критическим увеличением в кровеносном русле незрелых форм. Со временем происходит распространение патологических форм клеток во все органы и системы организма, что вызывает прогрессирование болезни.
На данном этапе однозначные факторы, приводящие к развитию заболевания, не определены. Существует несколько версий возникновения патологического состояния:

Кроме того, на появление опухолевого процесса влияют пол, возраст пациента и воздействие радиационного облучения в зоне проживания.
Проявление симптоматики опухолевого процесса крови зависит от формы болезни. Наиболее распространенная форма - хронический миелоидный лейкоз. Это состояние имеет злокачественный характер.
Хронический миелолейкоз - что это такое? Это состояние, которое возникает вследствие того, что в организме появляется аномальный ген, который поражает кровяные клетки. Место локализации гена - костный мозг. С током крови происходит распространение патологических клеток по всем органам.
Заболевание не имеет острого начала и яркой клинической картины. Для него характерно медленное течение. Опасность состоит в том, что эта форма болезни может перейти в острую фазу в любой период, что может закончиться летальным исходом для пациента.
Миелоидный лейкоз имеет несколько стадий развития:
Большинству пациентов ставят диагноз именно в этой стадии. Начало заболевания точно определить невозможно, поскольку оно имеет бессимптомный характер или легкие проявления. Сначала появляется утомляемость, тяжесть в желудке или в левом подреберье, одышка.

Во время приема пищи пациенты жалуются на чувство переполненности в эпигастрии. Пальпаторно ощущается увеличение селезенки. Осмотр сопровождается болезненными ощущениями со стороны селезенки, иррадиирущими в спину. В анализе крови определяется лейкоцитоз, нарастающий в динамике, а также тромбоцитоз и увеличение количества гранулоцитов.
Часто больные обращаются к врачу при развитии инфаркта селезенки. Появляется резкий болевой синдром в ее проекции, симптомы интоксикации организма, повышается температура тела.
В этой стадии заболевание практически не имеет проявлений. У больного нет жалоб, кроме периодического подъема температуры до субфебрильных показателей и усталости. Продолжает увеличиваться уровень миелоцитов и лейкоцитов в крови.
Уровень базофилов увеличивается на треть. После этого больных начинает беспокоить чувство жара и желание чесаться. Это связано с увеличением выработки гистамина.
Развитие третьей стадии описывает клиническую картину, схожую с острым течением болезни. Хронический миелоидный лейкоз прогрессирует, и появляется яркая клиническая картина. Пациенты жалуются на такие проявления:
При обследовании пациента можно обнаружить увеличение различных групп лимфатических узлов, печени, селезенки, развитие Бластный криз - финальная стадия болезни, которая характеризуется следующими клиническими проявлениями:

На этой стадии заболевания жизнь пациента зависит исключительно от уровня паллиативной терапии.
Клиническая картина развивается быстро, имеет яркие признаки заболевания. Без назначения адекватной терапии результат может быть неблагоприятным уже по истечении нескольких недель или месяцев.
Острый миелолейкоз - что это такое? Это злокачественный опухолевый процесс миелоидного ростка крови. Больные клетки не способны противостоять инфекциям, хотя это является их основной функцией. Параллельно с увеличением бластных структур происходит уменьшение остальных форменных элементов крови.
Эритропения и недостаток гемоглобина проявляются бледностью кожных покровов, одышкой, усталостью. Снижение количества тромбоцитов приводит к увеличению склонности кожи к повреждениям, повышенной кровоточивости, появлению петехий и гематом.
Первые симптомы не являются специфичными. Очень легко перепутать их с проявлениями респираторной вирусной инфекции. Кроме того, острый миелолейкоз сопровождается прогрессированием болевых ощущений в костях и суставах.
Успешность начала лечения зависит от скорости проведения диагностики и постановки правильного диагноза. Чтобы определить общее состояние и фазу заболевания пациента, у которого подозрение на миелолейкоз, анализы проводятся в следующих направлениях:

Миелоидный лейкоз требует безотлагательного начала лечения. Врач-онколог определяет схему терапии, исходя из стадии заболевания, его проявлений. На ранней стадии назначается витаминная диета, общеукрепляющие препараты.
Лечение миелолейкоза основывается на применении препаратов, способных угнетающе действовать на онкоген. Основные средства:
Также для укрепления иммунной системы пациентам назначается прием «Интерферона». Препарат не способен самостоятельно справиться с заболеванием, однако его применяют в комплексной терапии в виде ежедневных подкожных инъекций.
Химиотерапия проводится с использованием цитостатических средств. Эту часть терапии используют как дополнительное лечение при трансплантации клеток костного мозга. Эффективными считаются «Гидроксикарбамид», «Бусульфан», «Винбластин», «Винкристин», «Цитарабин».

Облучение при онкологии проводится при помощи высокоэнергетических лучей, а также их частиц. Применяется индивидуально, в зависимости от необходимости. При миелолейкозе лучевая терапия используется для уменьшения болевых синдромов в костях и суставах. Также облучение при онкологии кроветворной системы используют перед трансплантацией костного мозга.
Хирургическое вмешательство является довольно распространенным методом лечения, но при этом дорогостоящим. Не каждый пациент в состоянии себе это позволить. Онкоцентр на Каширке - один из знаменитых институтов терапии опухолевых новообразований - проводит подобные оперативные вмешательства, помогая излечиться своим пациентам.
Пересадка самого костного мозга сейчас используется не так часто, как которые берут из периферической крови. Существуют два варианта проведения процедуры:
Онкоцентр на Каширке не только проводит хирургические вмешательства, которые позволили снизить смертность пациентов, но и использует современные методы термоаблации, криотермоаблации и радиоволновой хирургии.

В статье был рассмотрен термин «миелолейкоз». Что это такое, вам теперь известно. Благоприятный исход возможен при полном курсе лечения начальных стадий заболевания. Терминальная стадия предполагает исключительно паллиативную терапию. Поздние и злокачественные стадии болезни приводят к летальному исходу у пациентов.
Диагноз (ХМЛ) в большинстве случаев установить или, во всяком случае, заподозрить нетрудно по характерным изменениям картины крови. Эти изменения выражаются в постепенно нарастающем лейкоцитозе, небольшом в начале заболевания (10- 15 10 9 /л) и достигающем по мере течения болезни без лечения огромных цифр - 200-500-800 10 9 /л и даже более.
Одновременно с нарастанием числа лейкоцитов отмечаются характерные изменения лейкоцитарной формулы: увеличение содержания гранулоцитов до 85-95 %, наличие незрелых гранулоцитов - миелоцитов, метамиелоцитов, при значительном лейкоцитозе - нередко промиелоцитов, а иногда единичных бластных клеток. Очень характерно увеличение содержания базофилов до 5-10 %, нередко с одновременным повышением уровня эозинофилов до 5-8 % («эозинофильно-базофильная ассоциация», не встречающаяся при других заболеваниях) и уменьшением числа лимфоцитов до 10-5 %.
Иногда количество базофилов достигает значительных цифр- 15-20 % и более.
В литературе 15-20-летней давности в таких случаях заболевание обозначалось как базофильный вариант хронического миелолейкоза, который встречается у 5-8 % больных. Описан эозинофиль-ный вариант, при котором в крови постоянно 20- 40 % эозинофилов. В настоящее время эти варианты не выделяют, а увеличение числа базофилов или эозинофилов рассматривается как признак продвинутой стадии болезни.
У большинства больных увеличено количество тромбоцитов до 400-600 10 9 /л, а иногда и более - до 800-1000 10 9 /л, редко - еще выше. Содержание гемоглобина и эритроцитов может долго оставаться нормальным, снижаясь только при очень высоком лейкоцитозе. У некоторых больных в начале заболевания наблюдается даже небольшой эритроцитоз - 5,0-5,5 10 12 л.
Исследование костно-мозгового пунктата обнаруживает увеличение числа миелокариоцитов и процента незрелых гранулоцитов с увеличением миелоидно/эритроидного соотношения до 20-25/1 вместо нормального 3-4/1. Обычно увеличено количество базофилов и эозинофилов, особенно у больных с высоким содержанием этих клеток в крови. Как правило, отмечается большое количество фигур митоза.
У некоторых больных, чаще при значительном гиперлейкоцитозе , в костно-мозговом пунктате обнаруживаются голубые гистиоциты и клетки, напоминающие клетки Гоше. Это макрофаги, захватывающие глюкоцереброзиды из распадающихся лейкоцитов. Число мегакариоцитов обычно увеличено, как правило, они имеют признаки дисплазии.
При морфологическом исследовании не обнаруживается каких-либо изменений в строении клеток гранулоцитарного ряда при ХМЛ по сравнению с нормальными, однако при электронной микроскопии выявляется асинхронизм в созревании ядра и цитоплазмы: на каждом этапе созревания гранулоцита ядро отстает в своем развитии от цитоплазмы.
Из цитохимических особенностей очень характерно резкое снижение или полное исчезновение щелочной фосфатазы в нейтрофилах крови и костного мозга.
При трепанобиопсии
обнаруживаются выраженная гиперплазия миелоидного ростка, резкое уменьшение содержания жира, у 20-30 % больных уже в начале заболевания - та или иная степень миелофиброза.
Морфологическое исследование селезенки
обнаруживает инфильтрацию красной пульпы лейкемическими клетками.
Из биохимических изменений характерным является увеличение содержания витамина В12 в сыворотке крови, которое превышает нормальное иногда в 10-15 раз и нередко остается повышенным при клинико-гематологической ремиссии. Другое существенное изменение - увеличение содержания мочевой кислоты. Оно оказывается высоким практически у всех нелеченых больных при значительном лейкоцитозе и может повышаться еще больше при проведении цитостатической терапии.
У некоторых больных постоянное повышение уровня мочевой кислоты приводит к образованию уратовых мочевых камней и подагрических артритов, отложению кристаллов мочевой кислоты в тканях ушных раковин с образованием видимых узелков. У подавляющего числа больных отмечается высокий уровень лактатдегидрогеназы сыворотки.
Начало заболевания в большинстве случаев почти или совсем бессимптомно. Обычно при уже появившихся изменениях крови селезенка не увеличена. По мере развития болезни она прогрессивно увеличивается, иногда достигая огромных размеров. Лейкоцитоз и размеры селезенки не всегда коррелируют между собой. У некоторых больных селезенка занимает всю левую половину живота, спускаясь в малый таз, при лейкоцитозе 65-70 10 9 /л, у других больных с лейкоцитозом, достигающим 400-500 10 9 /л, селезенка выступает из-под края реберной дуги всего на 4-5 см. Большие размеры селезенки особенно характерны для ХМЛ с высокой базофилией.
При выраженной спленомегалии обычно увеличена и печень, но всегда в значительно меньшей степени, чем селезенка. Увеличение лимфатических узлов для ХМЛ не характерно, оно встречается иногда в терминальной стадии болезни и обусловлено инфильтрацией лимфатического узла бластными клетками.

Жалобы на слабость, чувство тяжести, иногда боли в левом подреберье, потливость, субфебрильную температуру появляются только при развернутой клинической и гематологической картине заболевания.
У 20-25 % больных ХМЛ выявляется случайно, когда еще нет клинических признаков болезни, а имеются лишь нерезко выраженные гематологические изменения (лейкоцитоз и небольшой процент незрелых гранулоцитов в крови), которые обнаруживают при анализе крови, сделанном по поводу другого заболевания или при профилактическом обследовании. Отсутствие жалоб и клинических симптомов иногда приводит к тому, что характерные, но умеренные изменения крови, к сожалению, не привлекают внимания врача, и истинное начало заболевания удается установить лишь ретроспективно при обращении больного с уже выраженной клинико-гематологической картиной болезни.
Подтверждением диагноза ХМЛ является обнаружение в клетках крови и костного мозга характерного цитогенетического маркера - Ph-хромосомы. Этот маркер имеется у всех больных ХМЛ и не встречается при других заболеваниях.
Хронический миелолейкоз - первое онкологическое заболевание, при котором у человека были описаны специфические изменения хромосом и расшифрованы молекулярные механизмы, лежащие в основе развития болезни.
В 1960 г. два цитогенетика из г. Филадельфии в США P. Nowell и D. Hungerford у всех обследованных ими больных ХМЛ обнаружили укорочение длинного плеча одной из, как они ошибочно считали, хромосом 21-й пары. По названию города, где было сделано открытие, эта хромосома была названа филадельфийской, или Ph-хромосомой. В 1970 г., используя более совершенную технику окрашивания хромосом, Т. Caspersson и соавт. установили, что при ХМЛ имеется делеция длинного плеча одной из хромосом, не 21-й, а 22-й пары. Наконец, в 1973 г. было сделано важнейшее открытие, ставшее отправной точкой в исследовании патогенеза ХМЛ: J. Rowley показала, что образование Ph-хромосомы обусловлено реципрокной транслокацией (взаимный обмен частью генетического материала) между хромосомами 9 и 22.
При такой транслокации происходит перенос большей части длинного плеча хромосомы 22 на длинное плечо хромосомы 9, а маленькой терминальной части длинного плеча хромосомы 9 - на хромосому 22. В результате возникает характерная цитогенетическая аномалия - удлинение длинного плеча одной из хромосом 9-й пары и укорочение длинного плеча одной из хромосом 22-й пары. Именно эта хромосома из 22-й пары с укороченным длинным плечом и обозначается как Ph-хромосома.
К настоящему времени установлено, что Ph-хромосома - t(9;22)(q34;q11) обнаруживается в 95- 100 % метафаз у 90-95 % больных ХМЛ. Примерно в 5 % случаев выявляются вариантные формы Ph-хромосомы. Чаще всего это сложные транслокации, вовлекающие хромосомы 9, 22 и какую-либо третью хромосому, а иногда добавочно 2 или 3 хромосомы. При сложных транслокациях всегда имеются такие же молекулярные изменения, как при стандартной t(9;22)(q34;q11). Стандартные и вариантные транслокации могут одновременно обнаруживаться у одного и того же больного в разных метафазах.

Иногда встречается так называемая маскированная транслокация с такими же, как при типичной, молекулярными изменениями, но не определяемая обычными цитогенетическими методами. Это обусловлено переносом меньших, чем при стандартной транслокации, участков хромосом. Описаны также случаи, когда при обычном цитогенетиче-ском исследовании не обнаруживается t(9; 22), однако методом FISH или RT-PCR (ПЦР в реальном времени) удается установить, что в типичном участке хромосомы 22 имеется стандартная для ХМЛ перестройка генов - образование химерного гена BCR-ABL. Исследования таких случаев показали, что иногда происходит перенос участка хромосомы 9 на хромосому 22, но отсутствует транслокация участка хромосомы 22 на хромосому 9.
В начальном периоде цитогенетического изучения хронического миелолейкоза выделяли два его варианта - Ph-позитивный и Ph-негативный. Впервые Ph-негативный ХМЛ описан S. Krauss и соавт. в 1964 г.. Авторы обнаружили Ph-негативный ХМЛ почти у половины больных, которых они наблюдали. В дальнейшем по мере совершенствования методов исследования доля Ph-негативного ХМЛ неуклонно сокращалась. В настоящее время признается, что истинного Ph-негативного (BCR-ABL-негативного) ХМЛ не существует, а описанные ранее наблюдения в большинстве случаев относились к BCR-ABL-позитивному ХМЛ, но с таким типом хромосомных перестроек, которые не могли быть выявлены известными в то время цитогенетическими методами.
Таким образом, полученные к настоящему времени данные позволяют считать, что во всех случаях ХМЛ существуют изменения хромосом 9 и 22 с одинаковой перестройкой генов в определенной области хромосомы 22. В тех случаях, когда характерных цитогенетических изменений не удается обнаружить, речь идет о других заболеваниях, похожих на ХМЛ по клиническим проявлениям (спленомегалия) и картине крови (гиперлейкоцитоз, нейтрофилез). Чаще всего это хронический миеломоноцитарный лейкоз (ХММЛ), который в классификации ВОЗ 2001 г. относится к болезням, имеющим как миелопролиферативные, так и миелодиспластические черты. При ХММЛ всегда повышено количество моноцитов в крови и костном мозге.
При хроническом миелолейкозе у многих больных обнаруживаются транслокации с участием хромосомы 5: t(5;7), t(5;10), t(5;12), при которых образуются слитные гены, вовлекающие расположенный на хромосоме 5 ген PDGFbR (ген b-рецептора ростового фактора, продуцируемого тромбоцитами, - platelet-derived growth factor receptor b). Продуцируемый этим геном белок имеет домен с функцией тирозинкиназы, активирующейся при транслокации, чем обусловлен нередко значительный лейкоцитоз.
При наличии лейкоцитоза , нейтрофилеза и молодых форм гранулоцитов в крови, дисплазии всех ростков миелопоэза, но отсутствии моноцитоза заболевание, согласно классификации ВОЗ, обозначают как атипичный ХМЛ, также рассматриваемый в рубрике миелодиспластических/миелопролиферативных болезней. В 25-40 % случаев это заболевание, как и другие формы миелодиспластических синдромов, заканчивается острым лейкозом. Характерных цитогенетических изменений не обнаруживается.
2707 0
Хронический миелоидный лейкоз (ХМЛ) является неопластическим клональным заболеванием мультипотентной гемопоэтической стволовой клетки с преимущественным вовлечением гранулоцитарной клеточной линии.
Заболевание впервые было описано Р. Вирховым в середине XIX века под названием «селезеночная лейкемия». На долю ХМЛ приходится примерно 20% всех лейкозов в Европе.
Чаще заболевают люди среднего и пожилого возраста с медианой возраста около 50 лет, хотя ХМЛ может развиваться в любом возрасте.
Нет зависимости в заболеваемости от половой и этнической принадлежности.
Этиология ХМЛ неизвестна. Среди выживших после атомных бомбардировок в Японии повышение заболеваемости ХМЛ наблюдалось после трехлетнего латентного периода с достижением пика через 7 лет. В группе пациентов в Великобритании, которым проводили лучевую терапию по поводу анкилозирующего спондилита, отмечено повышение заболеваемости хроническим миелоидным лейкоз после латентного периода в 13 лет.
В целом воздействие ионизирующего излучения отмечено в анамнезе менее чем у 5% больных ХМЛ. Контакт с миелотоксическими агентами выявлен в единичных случаях. Хотя при ХМЛ отмечено повышение частоты экспрессии антигенов HLA-Cw3 и HLA-Cw4, нет сообщений о случаях семейного ХМЛ. Заболеваемость ХМЛ составляет 1,5 на 100000 населения.
В 1960 г. G.Nowell и D.Hungerford обнаружили у больных ХМЛ укорочение длинного плеча одной хромосомы (Хр), как они считали, 21-й пары. Эта хромосома ими была названа филадельфийской, или Ph-хромосомой.
Однако в 1970 г. T.Caspersson и соавт. установили, что при хроническом миелоидном лейкозе имеется делеция одной из Хр 22-й пары. В 1973 г. J.Rowley показала, что образование Ph-хромосомы обусловлено реципрокной транслокацией (взаимным переносом части генетического материала) между Хр9 и Хр22. Эта измененная хромосома из 22-й пары с укороченным длинным плечом и обозначается как Ph-хромосома.
В начальном периоде цитогенетического изучения ХМЛ были описаны два варианта - Ph+ и Ph-. Однако теперь следует признать, что Ph- ХМЛ не существует, а описанные случаи, вероятно, относились к миелодиспластическим состояниям. Ph-хромосома, t (9; 22) (q34; q11) обнаруживается у 95-100% больных ХМЛ.
В остальных случаях возможно наличие следующих вариантов транслокации:
Сложные транслокации, вовлекающие Хр9, 22 и какую-либо третью хромосому,
- маскированные транслокации с такими же молекулярными изменениями, но не определяемыми обычными цитогенетическими методами,
- наличие t (9; 22) без переноса участка Хр22 на Хр9.
Таким образом, во всех случаях ХМЛ существуют изменения Хр9 и Хр22 с одинаковой перестройкой генов в определенной области Хр22 (2).
На длинном плече Хр9 (q34) расположен протоонкоген ABL (Абельсона), который кодирует через синтез специфической мРНК образование белка р145 , относящегося к семейству тирозинкиназ (ТК) - энзимов, катализирующих процессы фосфорилирования аминокислот в клеточном цикле. На длинном плече Хр22 (q 11) расположен район M-BCR (Major breakpoint cluster region).
Ген, расположенный в этом районе, обозначается как ген BCR. Он кодирует образование белка p160BCR, который участвует в регуляции некоторых функций нейтрофилов. В результате транслокации t(9;22)(q34;q11) протоонкоген с-аcr переносится в район bcr Хр22.
Обычно разрыв гена BCR происходит между экзонами b2 и b3 или экзонами b3 и b4, причем происходит слияние экзона 2 гена ABL с оставшейся на Хр22 частью гена BCR (с экзоном b2 или b3). В результате этого формируется химерный ген BCR-ABL, кодирующий аномальную 8,5кб рибонуклеиновую кислоту (мРНК) , которая продуцирует фузионный протеин p210BCR-ABL, обладающий тирозинкиназной активностью.
Иногда точка разрыва гена BCR находится в районе m-BCR (minor breakpoint cluster region), при этом продукцией химерного гена является 7,5 кб мРНК, кодирующая белок p190BCR-ABL. Такой тип транслокации ассоциирован с вовлечением в процесс клеток лимфоидной линии и часто обуславливает развитие Ph+ острого лимфобластного лейкоза (ОЛЛ) .
В связи с активацией гена ABL, возникающей при его слиянии с геном BCR, белок р210BCR-ABL имеет значительно более выраженную тирозинкиназную активность, чем его нормальный прототип p145ABL. ТК фосфорилируют тирозин в белках, которые регулируют рост и дифференцировку клеток, в том числе и гемопоэтических.
Мутации тирозинкиназ с повышением их активности приводят к нерегулируемому фосфорилированию тирозина и, соответственно, к нарушению процессов роста и дифференцировки клеток. Однако это не единственный и не главный механизм в патогенезе симптомов ХМЛ.
Биологический эффект химерного гена BCR-ABL сводится к следующим основным нарушениям в жизнедеятельности клетки:
Увеличению митогенной активности в связи с усилением передачи сигнала пролиферации путем активации рецепторов гемопоэтических клеток благодаря усилению фосфорилирования. Это не только усиливает пролиферацию, независимо от регулирующего влияния ростовых факторов, но и нарушает дифференцировку клеток-предшественников;
Нарушению адгезии клеток к строме, что ведет к снижению времени взаимодействия строма/гемопоэтические клетки. Следствием этого является нарушение нормальной последовательности пролиферация/созревание, поэтому клетки-предшественники находятся дольше в поздней прогениторной пролиферативной фазе перед дифференцировкой. Это приводит к увеличению пролиферации и времени циркуляции клеток-предшественников и появлению очагов экстрамедуллярного кроветворения;
Ингибированию апоптоза вследствие защитного действия белка р210 и активации гена MYC, являющегося ингибитором апоптоза, а также вследствие гиперэкспрессии гена BCL-2. Вследствие этого лейкоциты при ХМЛ живут дольше, чем нормальные клетки. Характерной особенностью белка p210BCR-ABL является способность к аутофосфорилированию, приводящей к автономной активности клетки и практически полной ее независимости от внешних регуляторных механизмов;
Возникновению нестабильного генома клетки в связи со снижением функции гена ABL, поскольку при его делеции снижается его роль как супрессора опухолевого роста. Вследствие этого не происходит остановки клеточной пролиферации. Кроме того, в процессе пролиферации активируются другие клеточные онкогены, что ведет к дальнейшему усилению пролиферации клеток.
Итак, усиление пролиферативной активности, снижение чувствительности к апоптозу, нарушение процессов дифференцировки, повышенная способность незрелых гемопоэтических клеток-предшественников к выходу из костного мозга в периферическую кровь являются основными характеристиками лейкемических клеток при хроническом миелоидном лейкоз.
Хроническая фаза (ХФ) болезни в большинстве случаев протекает почти или вовсе бессимптомно. Жалобы на повышенную утомляемость, слабость, иногда тяжесть в эпигастрии. При обследовании можно обнаружить увеличение селезенки и крайне редко - печени.
Клинико-гематологическая картина может быть бессимптомной, количество лейкоцитов и тромбоцитов может быть нормальным или незначительно увеличенным; в лейкоцитарной формуле может наблюдаться умеренный левый сдвиг - единичные метамиелоциты и миелоциты, иногда небольшое повышение количества базофилов. При цитологическом исследовании выявляется лишь Ph-хромосома без дополнительных изменений со стороны других хромосом.
В фазе акселерации пациенты отмечают повышенную утомляемость при выполнении привычной работы, дискомфорт в левом подреберье; потеря веса, периодические «немотивированные» повышения температуры тела отражают наличие гиперкатаболизма. Как правило, определяется увеличение селезенки и в 20-40% случаев - увеличение печени.
Основным признаком перехода заболевания в ФА являются изменения в анализах крови: нарастает неконтролируемый цитостатическими препаратами лейкоцитоз с количественным преобладанием незрелых форм лейкоцитов, возрастает количество базофилов, реже нарастает количество эозинофилов или моноцитов.
Количество тромбоцитов может увеличиваться с развитием тромботических осложнений в начале ФА с последующим развитием тромбоцитопении с проявлениями геморрагического синдрома по петехиально-пятнистому типу. В костном мозге в ФА выявляется некоторое увеличение количества бластных клеток (обычно менее 20%) и увеличение содержания промиелоцитов и миелоцитов. При цитогенетическом исследовании в ФА, помимо наличия Ph-хромосомы, можно выявить добавочные изменения других хромосом, что свидетельствует о появлении более злокачественного клеточного клона.
В фазе бластного криза появляются резкая общая слабость, выраженные оссалгии за счет поднадкостничной инфильтрации бластными клетками, периодическая лихорадка, потливость, выраженное снижение массы тела. Нарастает гепатоспленомегалия. Как правило, отмечается выраженный геморрагический диатез. Гематологические проявления характеризуются увеличением количества бластных клеток в периферической крови и/или костном мозге выше 20% при вариабильном количестве лейкоцитов.
Преобладающим по частоте вариантом БК является миелобластный вариант - примерно 50% всех случаев; лимфобластный и недифференцированный варианты - около 25% случаев каждый. Лимфобластный БК имеет чрезвычайно злокачественный характер, что связано с изменениями бластных клонов и в связи с этим с резистентностью к проводимой терапии.
Иногда БК характеризуется резким увеличением количества базофилов разной степени зрелости в периферической крови и костном мозге без большого количества бластных клеток. В некоторых случаях базофилия сменяется моноцитозом.
Обычно отмечается нормохромная анемия и тромбоцитопения разной степени тяжести, нормобластоз и фрагменты мегакариоцитов в мазке крови. Примерно у 10-15% больных в фазе БК появляются экстрамедуллярные бластные инфильтраты.
Реже наблюдаются поражения центральной нервной системы с симптомами нейролейкемии или поражение периферических нервов. У некоторых больных при БК бывают кожные лейкемиды или приапизм в результате лейкостаза и лейкозной инфильтрации пещеристых тел. Следует отметить, что в некоторых случаях при наличии экстрамедуллярных очагов бластной инфильтрации картина периферической крови и костного мозга может не иметь признаков перехода ХМЛ в фазу БК.
Согласно классификации ВОЗ (2002 г.), выделены следующие критерии для ФА и БК.
Фаза акселерации при наличии одного или более признаков:
Бласты 10-19% в периферической крови или костном мозге,
- базофилы менее 20% в периферической крови,
- персистирующая тромбоцитопения (менее 100,0х10 9 /л) или персистирующий тромбоцитоз более 1000,0х10 9 /л, несмотря на проводимую терапию,
- увеличение размеров селезенки и повышение уровня лейкоцитов, несмотря на проводимую терапию,
- цитогенетические данные в пользу клональной эволюции (в дополнение к цитогенетическим аномалиям, выявленным во время установления диагноза ХФ ХМЛ),
- мегакариоцитарная пролиферация в виде кластеров в сочетании со значительным ретикулиновым и коллагеновым фиброзом и/или выраженной гранулоцитарной дисплазией.
Фаза властного криза при наличии одного или более признаков:
Бласты 20% и более в периферической крови или костном мозге,
- экстрамедуллярная пролиферация бластов,
- большие скопления или кластеры бластов в костном мозге при трепанобиопсии.
Хроническая фаза ХМЛ устанавливается при отсутствии критериев ФА и фазы БК.
Спленомегалия и гепатомегалия любых размеров не являются признаками ФА и БКХМЛ.
Важно определять не только фазу ХМЛ, но и группу риска прогрессии болезни в дебюте заболевания с учетом данных первичного обследования пациента. J.E.Sokal и соавт. в 1987 г. предложили прогностическую модель с учетом четырех признаков: возраста больного в момент установления диагноза, размеров селезенки, числа тромбоцитов и числа бластов в крови. Эта модель получила наибольшее распространение и используется в большинстве исследований.
Расчет прогностического индекса проводится по формуле:
Индекс Sokal = exp(0,0116(возраст - 43,4) + 0,0345(размер селезенки - 7,51) + 0,188[(число тромбоцитов: 700)2 - 0,563] + 0,0887(число бластов в крови - 2,10)}.
Exp (экспонента) -2,718 возводится в степень того числа, которое получается в фигурных скобках.
При индексе менее 0,8 - группа низкого риска; при индексе 0,8-1,2 - группа среднего риска; при индексе более 1,2 - группа высокого риска.
К обязательным методам обследования пациентов для установления диагноза ХМЛ относятся:
Морфологическое исследование периферической крови с подсчетом лейкоцитарной формулы и количества тромбоцитов,
- морфологическое исследование пунктата костного мозга,
Поскольку единственным достоверным критерием диагноза хронический миелоидный лейкоз является наличие Ph- хромосомы, необходимо цитогенетическое исследование костного мозга с анализом не менее 20 метафазных пластинок; при негативном ответе - отсутствии t (9; 22) (q34; q11) - при высокой возможности диагноза ХМЛ необходимо использовать молекулярно-генетические методики-FISH (fluorescence in situ hybridization) или полимеразной цепной реакции (ПЦР)
,
- пальпаторное и УЗИ-определение размеров селезенки, печени, лимфатических узлов. Поскольку спленомегалия или гепа-томегалия любых размеров не являются критериями ФА или фазы БК, специфическое поражение любых других органов и тканей следует рассматривать как признак трансформации болезни в БК,
HLA- типирование для потенциальных кандидатов на аллогенную трансплантацию гемопоэтических стволовых клеток (алло-ТГСК)
показано больным ХМЛ в ФА и БК, не имеющим противопоказаний к использованию этого метода лечения,
- пациентам в фазе БК ХМЛ для определения типа бластов показано цитохимическое исследование и иммунофенотипирование.
К факультативным методам обследования относятся:
Трепанобиопсия для оценки наличия и распространенности процесса фиброза в костном мозге,
- инструментальные методы обследования - ультразвуковое исследование (УЗИ)
, магнитно-резонансная томография (МРТ)
, люмбальная пункция с целью определения наличия экстрамедуллярных очагов кроветворения,
- до начала терапии ингибиторами тирозинкиназы (ИТК)
целесообразно выполнение ПЦР для определения начального уровня экспрессии гена BCR-ABL.
Стандартная алло-ТГСК вызвала длительную молекулярную ремиссию или выздоровление у 50% больных со значительной разницей при учете групп риска. В странах, где доступна терапия ИТК и осуществляется алло-ТГСК, обе эти стратегии не являются взаимоисключающими, хотя после внедрения ИТК в клиническую практику заметно снижение ежегодного количества алло-ТГСК в последние 7 лет.
Эффективность проводимой терапии определяется по следующим критериям:
1. Наличие гематологической ремиссии: данные анализов крови:
- полная клинико-гематологическая ремиссия (CHR)
:
- тромбоциты ниже 450,0x10%,
- лейкоциты ниже 10,0x10%,
- в лейкограмме бласты менее 5%, отсутствуют незрелые гранулоциты.
2. Наличие цитогенетической ремиссии: наличие Ph хромосомы:
Полная - 0%,
- частичная - 1-35%,
- малая - 36-65%,
- минимальная - 66-95%.
3. Наличие молекулярной ремиссии: наличие BCR-ABL транскрипта:
Полная - транскрипт не определяется,
- большая - 0,1%.
Полная цитогенетическая (CCyR) и частичная цитогенетическая ремиссия (PCyR) в комбинации могут рассматриваться как большая цитогенетическая ремиссия (MCyR) . Большая молекулярная ремиссия (MMolR) является эквивалентом 1000-кратной редукции от базового уровня в 100%.
Полная молекулярная ремиссия (CMolR) констатируется, если транскрипт BCR-ABL не определяется методом RQ-PCR (real-time quantitative polymerase chain reaction).
С целью достижения циторедукции,
- при беременности для поддержания гематологического ответа,
- в случаях резистентности и/или непереносимости препаратов интерферона или ИТК,
- при невозможности выполнения алло-ТГСК,
- при невозможности обеспечения больных ХМЛ достаточным количеством ИТК.
Обычно терапия HU заключается в назначении данного препарата в дозе 2-3,0 грамма в сутки в сочетании с приемом аллопуринола в суточной дозе 600-800 мг при достаточной гидратации. Доза корригируется в зависимости от степени снижения уровня лейкоцитов, при снижении их ниже 10,0х10 9 /л переходят на прием поддерживающей дозы - 0,5 г/сутки с или без приема аллопуринола. Желательно количество лейкоцитов поддерживать на уровне не выше 6-8,0х10 9 /л.
В случае снижения количества лейкоцитов ниже 3,0х10 9 /л прием препарата временно прекращается. Переносимость препарата достаточно хорошая, но при длительном применении возможно образование язв желудка.
Внедрение в практику препаратов rINF позволило получить у части больных ХМЛ не только длительную клинико-гематологическую, но и цитогенетическую ремиссии, хотя частота полного цитогенетического ответа (CCyR) была низкой - 1015%. Сочетание препаратов rINF+LDAC несколько увеличило частоту CCyR (25-30%), однако рано или поздно практически у всех больных этой группы заболевание прогрессировало.
1-я неделя: по 3 млн. Ед/м2 подкожно ежедневно,
- 2-я и 3-я недели: по 5 млн. Ед/м подкожно ежедневно,
- в дальнейшем препарат назначают по 5 млн ЕД/м подкожно ежедневно или 3 раза в неделю.
Препарат может вызывать аллергические реакции, повышение температуры тела, зуд кожи, боли в мышцах (обычно в начале применения). Терапия обычно продолжается в течение 2-х лет, в дальнейшем наблюдается уход из-под контроля препарата.
При комбинации rINF+LDAC (цитозар в дозе 20 г/м2 подкожно 2 раза в день в течение 10 дней ежемесячно) цитогенетический ответ был выше, чем при терапии только rINF, но различий в общей выживаемости не было.
Сравнение результатов применения rINF в дозе 3 млн. Ед/м 3 раза в неделю и в дозе 5 млн. ЕД/м ежедневно показало, что малые дозы такие же по эффективности, как и высокие дозы, но переносятся лучше. Однако у всех пациентов, находящихся на такой терапии, определялось наличие минимальной остаточной болезни, что предполагает неизбежность развития рецидива.
В рутинной клинической практике последовательное или сочетанное применение ИМ или новых ИТК с препаратами rINF пока не рекомендуется, поскольку неизвестны результаты проводимых клинических исследований. В настоящее время применение rINF можно рекомендовать в тех же случаях, в которых рекомендуется терапия гидроксимочевиной.
Проведение алло-ТГСК в качестве терапии первой линии при наличии донора, совместимого по системе HLA, а также возраста больного ниже 50-55 лет, с начала 90-х годов 20-го века стало стандартной рекомендацией для пациентов с первично диагностированным ХМЛ. Алло-ТГСК считается единственным методом, способным полностью элиминировать из организма лейкозный клон клеток.
Тем не менее, существует несколько проблем, которые ограничивают ее широкое применение у больных ХМЛ:
Преобладание в популяции больных ХМЛ возрастной группы 50-60 лет,
- невозможность для большинства пациентов найти HLA-совместимого родственного или неродственного донора,
- летальность до 20% в раннем посттрансплантационном периоде от осложнений полихимиотерапии (ПХТ)
или реакции «трансплантат против хозяина» (РТПХ)
.
В ФА решение о проведении алло-ТГСК должно приниматься с учетом следующих данных:
Оценка степени риска прогрессии хронического миелоидного лейкоза (по индексу Sokal),
- определение эффективности ИТК с учетом цитогенетики и данных ПЦР,
- оценка риска трансплантационных и посттрансплантационных осложнений,
- наличие доступного донора.
Согласно рекомендациям ЕВМТ, при ХМЛ выполнение алло-ТГСК в ХФ, в ФА или в поздней ХФ показано от родственного или неродственного совместимого донора, не показано от неродственного несовместимого донора; проблема выполнения ауто-ТГСК находится в стадии разработки. В фазе БК выполнение алло- или ауто-ТГСК не показано.
Если принято решение о выполнении алло-ТГСК, встает вопрос о том, какой режим кондиционирования предложить пациенту: миелоаблативный или немиелоаблативный. Одним из миелоаблативных режимов при проведении алло-ТГСК у пациентов ХМЛ является BuCy: бусульфан в дозе 4 мг/кг веса в день и циклофосфан 30 мг/кг веса в день в течение 4-х дней до алло-ТГСК.
Немиелоаблативный (редуцированный) режим Bu-Flu-ATG состоит из однократного введения комбинации бусульфана в дозе 8 мг/кг веса, флюдарабина 150мг/м и кроличьего антитимоцитарного глобулина в дозе 40 мг. Однако в связи с отсутствием рандомизированных исследований не рекомендуется применять этот вариант в качестве стандарта лечения.
Осознание роли тирозинкиназной активности (ТКА) белка BCR-ABL в процессе миелопролиферации привело к синтезу новой серии препаратов, таргентных в отношении кодируемых BCR-ABL протеинов. Ингибирование ТКА приводит к прерыванию сигналов, контролирущих лейкемический фенотип. Первый из ингибиторов ТКА, иматиниб мезилат (ИМ), обладает высокой и относительно специфической биохимической активностью при ХМЛ, что привело к его быстрому внедрению в клиническую практику.
С появлением ИТК показания к алло-ТГСК резко изменились. В ранней ХФ ХМЛ алло-ТГСК показана при развитии резистентности или непереносимости к ИТК, поэтому ее выполнение взрослым пациентам в качестве терапии первой линии на сегодняшний день не рекомендовано.
Однако из этого правила есть два исключения:
В педиатрической практике предпочтительнее в качестве первичной терапии применять алло-ТГСК при наличии HLA-совместимого родственного донора,
- если стоимость предполагаемого лечения ИТК значительно превышает стоимость алло-ТГСК.
В целом, большинству пациентов с ХМЛ в ХФ, если возможно, рекомендуется проводить начальную терапию ИМ.
Иматиниб мезилат (ИМ) - гливек, являющийся ингибитором тирозинкиназы, применен в клинике в 1995 г. ИМ (2-фениламинопиримидин) эффективно блокирует киназную активность белка BCR-ABL и может блокировать другие белки с протеинкиназной активностью, необходимые для нормального выживания клетки.
Исследования показали, что ИМ избирательно ингибирует пролиферацию клеток при хроническом миелоидном лейкозе. Препарат преимущественно элиминируется печенью, 50% снижение его концентрации в плазме составляет около 18 часов. Рекомендуемая начальная доза препарата составляет 400 мг/день, что позволяет достичь п олной клинико-гематологической ремиссии (CHR) в 95% и CCyR в 76% случаев. В группе пациентов с CCyR большая молекулярная ремиссия (MMolR) определялась только в 57% случаев.
Применение ИМ в «поздней» ХФ в такой же дозировке позволяет достичь CCyR в 41-64% со свободной от прогрессирования выживаемсти в 69% больных. При применении ИМ в ФА в дозе 600 мг/день CHR была достигнута в 37%, CCyR в 19% случаев и трехлетняя PFS у 40% больных. При применении ИМ в той же дозе в БК ХМЛ CHR была достигнута в 25%, PFS составила менее 10 месяцев, общая выживаемость свыше 3-х лет - в 7% случаев.
Поскольку частота CCyR очень высока у пациентов при лечении ИМ, необходимо измерение уровня BCR-ABL транскрипта для определения наличия минимальной остаточной болезни (МОБ) . Частота отсутствия данного транскрипта рассматривается как CMolR, весьма вариабельна и колеблется в пределах 4-34%.Показано, что Ph+ стволовые клетки менее чувствительны к ИМ, чем поздние Ph+ прогениторы.
В случае субоптимального эффекта от применения ИМ в ХФ в дозе 400 мг/день предлагается эскалация дозы препарата до 600-800 мг/день при условии, что резистентность к ИМ не связана с дополнительными мутациями BCR-ABL. Прием ИМ в дозе 600 мг в сутки значительно более эффективен в ФА и БК. У пациентов в ХФ с гематологической и цитогенетической резистентностью к ИМ в дозе 400 мг/день повышение дозы ИМ до 800 мг в день обусловило CHR у 65% и CCyR - у 18% пациентов.
При применении ИМ могут наблюдаться некоторые осложнения:
Анемия и/или панцитопения,
- инфраорбитальный отек, редко - генерализованный отек,
- боли в костях и суставах,
- снижение уровня кальция и фосфора в крови,
- зуд кожи.
На сегодняшний день существует два препарата группы ИТК, зарегистрированные к применению в качестве препаратов 2-й линии терапии ХМЛ в случаях развития резистентности к ИМ: дазатиниб и нилотиниб.
Дазатиниб (спрайсел) является ингибитором ABL-киназ (всего ингибирует около 50 киназ) и отличается от ИМ тем, что может связывать как активные, так и неактивные (открытые и закрытые) конформации киназного ABL-домена, а также ингибирует семейство киназ Src, включая Srk и Lyn.
Его можно рассматривать как двойной ингибитор. Дазатиниб в 300 раз более активен, чем ИМ, и к тому же активен против большинства ИМ-резистентных мутантных субклонов, за исключением клона T315I и, вероятно, мутантного клона F317L. Препарат применяется для лечения пациентов ХМЛ при резистентности или непереносимости ИМ. Ремиссия наблюдалась в одинаковой степени у пациентов при наличии и при отсутствии мутаций киназы, кроме мутаций T315I.
Препарат может вызывать осложнения в виде нейтропении, тромбоцитопении, рвоты, диареи, желудочно-кишечных кровотечений, генерализованного отека, кожных высыпаний, гипертензии, ХОБЛ. У единичных пациентов может наблюдаться плевральный и перикардиальный выпот. Для коррекции осложнений следует сделать перерыв в приеме препарата, назначить диуретики, кортикостероиды, при необходимости - торакоцентез.
Доза препарата 100 мг один раз в день сопоставима по эффективности с дозой 70 мг два раза в день, но обладает лучшей переносимостью.
Нилотиниб (тасигна) является дериватом аминопиримидина, т.е. модифицированным производным ИМ, что объясняет их схожий спектр ингибирования (ингибирует четыре ТК). Препарат обладает повышенной способностью связывать район АТФ онкопротеина BCR-ABL. Он в 20-50 раз более эффективен в сравнении с ИМ в отношении чувствительных к ИМ лейкемических клеток, а также активен в отношении всех ИМ-резистентных клеточных линий с мутациями киназного ABL-домена, за исключением мутации T315I и, вероятно, мутантного клона Y253H.
В группе больных в ХФ ХМЛ, резистентных к ИМ, достигнута CHR у 71% и CCyR - у 48% пациентов. Общая 2-летняя выживаемость в этой группе составила 95%. Не было различий в количестве ремиссий у пациентов при наличии или отсутствии мутации киназного ABL-домена. При применении препарата в ФА спустя один месяц после начала терапии в 55% случаев была зарегистрирована CHR, общая выживаемость спустя 12 месяцев составила 82%. В фазе БК при проведении терапии в течение 12 месяцев общая выживаемость составила 47%.
Кожный зуд,
- запоры,
- повышение уровня печеночных ферментов,
- повышение уровня непрямого билирубина,
- высыпания на коже.
Для дазатиниба 50% снижение уровня в плазме составляет 3-5 часов, для нилотиниба и ИМ - 15-18 часов. Для дазатиниба длительное ингибирование протеина BCR-ABL не означает обязательную элиминацию лейкемических клеток при хроническом миелоидном лейкозе . Поэтому постулат о превалировании эффективности длительного ингибирования киназ в лечении ХМЛ неприменим в отношении дазатиниба.
В общем, дазатиниб и нилотиниб обладают примерно равной активностью у пациентов при отсутствии эффекта от терапии ИМ. Однако ни один из них не рекомендуется для применения у пациентов с мутантным клоном N315I.
В стадии клинических испытаний находится препарат босутиниб, ингибирующий киназы ABL и Srk, и поэтому являющийся двойным ингибитором киназ. Он активен против клеточных линий, несущих мутации трех из четырех киназных доменов. Однако следует учитывать, что применение вышеуказанных препаратов не обеспечивает полного излечения.
После применения иматиниба в случае развития резистентности к препарату, при его непереносимости или выраженных осложнениях пациентам должна быть предложена терапия ИТК 2-й линии терапии;
- выбор препарата должен определяться степенью его токсичности.
Алло-ТГСК предлагается при:
Наличии мутаций T315I и других мутаций,
- отсутствии эффекта при лечении ИТК в ФА и БК,
- отсутствии эффекта при лечении ИТК 2-й линии терапии.
Хронический миелолейкоз
Хронический миелоидный лейкоз (ХМЛ) - медленно прогрессирующий тип лейкоза, характеризующийся неконтролируемым образованием клеток миелоидного ряда в костном мозге и выходом незрелых клеток в периферическую кровь.
Клетки, образующиеся при лейкозе, представляют собой аномальные незрелые формы. Продолжительность жизни этих незрелых клеток больше, чем зрелых лейкоцитов. По мере развития заболевания незрелые клетки накапливаются в костном мозге, вытесняя нормальные клетки кроветворения.
Причины ХМЛ
Возникновение ХМЛ почти всегда связано с мутацией гена в хромосоме, который называется Филадельфийской хромосомой. Эта мутация происходит постепенно в течение жизни. Она не передается от родителей к детям. В большинстве случаев причина мутации не известна. Исследования показывают, что на развитие ХМЛ влияет воздействие больших доз радиации, например, после ядерных аварий или атомных взрывов. Однако большинство больных ХМЛ не подвергались воздействию радиации.
Симптомы ХМЛ
Приведенные симптомы, кроме хронического миелолейкоза, могут быть вызваны другими, менее серьезными заболеваниями. Если вы испытываете любой из них, обратитесь за консультацией к врачу.
Слабость
Нехватка энергии
Усталость
Необъяснимая потеря веса
Ночные поты
Лихорадка
Боль или чувство полноты ниже ребер
Боль в костях
Боль в суставах
Уменьшение толерантности к физической нагрузке
Увеличение печени или селезенки
Беспричинные кровотечения или кровоподтеки.
Диагностика ХМЛ
Врач спросит о симптомах и истории болезни, а также выполнит медицинский осмотр. Врач может также проверить на наличие опухания печень, селезенку или лимфатические узлы в подмышечных впадинах, в паху или на шее. Вы можете быть направлены в онкологу - врачу, который специализируется на лечении рака.
Тесты могут включать:
Анализы крови - для проверки изменений в количестве или появлении различных типов клеток крови
Аспирация костного мозга - удаление образца жидкости костного мозга для проверки на наличие раковых клеток
Биопсия костного мозга - удаление образца жидкости костного мозга и небольшого образца кости для проверки на наличие раковых клеток
Осмотр образцов под микроскопом - исследование образцов крови, жидкости костного мозга, ткани лимфатических узлов или спинномозговой жидкости
Анализы костей, крови, костного мозга, ткани лимфатического узла или спинномозговой жидкости - чтобы классифицировать тип лейкемии и определить, есть ли лейкозные клетки в лимфатических узлах или спинномозговой жидкости
Цитогенетический анализ - тест, позволяющий найти определенные изменения в хромосомах (генетический материал) лимфоцитов. Используется, чтобы установить конкретный диагноз и разработать план лечения ХМЛ
Рентген грудной клетки - позволяет обнаружить признаки инфекции легких или рак груди
Компьютерная томография брюшной полости - вид рентгена, который использует компьютер, чтобы сделать снимки органов внутри тела
МРТ - тест, который использует магнитные волны, чтобы сделать снимки структуры внутри тела
УЗИ - обследование, которое использует звуковые волны для изучения внутренних органов.
Лечение ХМЛ
Методика лечения ХМЛ зависит от стадии заболевания и состояния здоровья пациента.
Лекарственная терапия хронического миелолейкоза
Разработаны лекарства, которые могут подавлять молекулы, провоцирующие развитие лейкемии и гена, связанного с ним. Эти препараты часто используют на ранних стадиях ХМЛ. Они являются лучшим вариантом лечения, чем химиотерапия и биологическая терапия. Для лечения хронического миелолейкоза применяются следующие лекарства:
Иматиниб (Гливек, Генфатиниб, Филахромин, Неопакс, Иматиниб и т.д.)
Дазатиниб (Спрайсел)
Нилотиниб (Тасигна).
Босутиниб (Bosutinibum).
Химиотерапия при хроническом миелолейкозе
Химиотерапия - использование лекарственных препаратов для уничтожения раковых клеток. Препараты для химиотерапии могут быть предоставлены в различных формах: таблетки, инъекции, введение через катетер. Лекарства поступают в кровоток и разносятся по всему телу, убивая главным образом раковые, а также некоторые здоровые клетки.
Биологическая терапия
Этот метод лечения ХМЛ все еще проходит испытания в клинических условиях. Лечение заключается в применении лекарств или веществ, которые используются для увеличения или восстановления естественной защиты организма от рака. Этот тип терапии также называется лечение с использованием модификаторов биологических реакций. Иногда используются очень специфические (моноклональные) антитела, разработанные для подавления лейкозных клеток. В настоящее время терапия с помощью моноклональных антител ограничена клиническими испытаниями и недоступна в России.
Химиотерапия с трансплантацией стволовых клеток
Химиотерапия с трансплантацией стволовых клеток для лечения ХМЛ все еще проходит клинические испытания. Проведение химиотерапии сопровождается трансплантацией стволовых клеток (незрелые клетки крови). Они заменят кроветворные клетки, разрушенные лечением рака. Стволовые клетки отбираются из крови или костного мозга донора, после чего вводятся в организм пациента.
Подсадка лимфоцитов
Лимфоциты - тип белых кровяных клеток. Лимфоциты от донора вводятся в организм пациента и раковые клетки не атакуют их.
Операция при хроническом миелолейкозе
Может быть проведена спленэктомия - операция по удалению селезенки. Она проводится, если селезенка увеличилась или появились другие осложнения.
ТРАНСПЛАНТАЦИЯ
Пересадка костного мозга
Из-за того, что химиотерапевтические препараты разрушают клетки костного мозга, пересадка является по-настоящему спасительным средством для пациента. Целью трансплантации костного мозга является внедрение в организм здоровых клеток костного мозга параллельно с лечением высокими дозами химиотерапевтических средств (тем самым повышается вероятность уничтожения раковых клеток и полного выздоровления).
Пересадка стволовых клеток
Стволовыми называют клетки, на ранних стадиях развития, еще не превратившиеся в лейкоциты, эритроциты или тромбоциты. Стволовые клетки в наши дни получают из периферической крови при помощи специального прибора, позволяющего рассортировать клетки разных типов. В таком приборе кровь центрифугируется с большой скоростью и разделяется на составные элементы. Процесс длится 3-4 часа.
Стволовые клетки отбираются и замораживаются до процедуры пересадки. Если трансплантация прошла успешно, стволовые клетки приживутся в организме реципиента, пройдут процесс созревания, и из них впоследствии образуются все виды клеток крови: лейкоциты, эритроциты и тромбоциты. Пересадка клеток от донора называется аллогенной трансплантацией, пересадка клеток самого пациента (как правило, стволовых) – аутологичной трансплантацией.
Аллогенная трансплантация (от совместимого донора)
При аллогенной трансплантации источником клеток костного мозга или стволовых клеток является донор, чьи клетки подобыли признаны подходящими для пересадки после анализа на тканевую совместимость. В некоторых случаях донором может стать родственник пациента, но в принципе можно использовать клетки постороннего человека, если они успешно прошли проверку на совместимость.
Перед процедурой трансплантации необходимо полностью уничтожить все злокачественные клетки в костном мозге пациента. Для этого назначаются цитотоксичные препараты в высоких дозах и радиотерапия (облучение всего организма). Затем трансплантат вводится в организм пациента через внутривенную инфузию.
Процесс приживания трансплантированных клеток занимает несколько недель. Все это время иммунная система пациента функционирует на крайне низком уровне, поэтому в этот период нужно тщательно оберегать больного от инфекций. По этой причине после процедуры пересадки пациент находится в изоляторе, пока в его анализе крови не будет отмечен рост числа лейкоцитов. Такой рост является симптомом восстановления иммунной системы, приживания трансплантата и возобновления процесса кроветворения.
В течение нескольких месяцев после процедуры пересадки важно оставаться под медицинским наблюдением, чтобы в случае необходимости вовремя распознать состояние, называемое «трансплантат против хозяина». Это состояние, при котором клетки пересаженного костного мозга атакуют ткани организма пациента. Оно может возникнуть в течение 6 месяцев после процедуры трансплантации. Реакция «трансплантат против хозяина» может сопровождаться симптомами различной степени тяжести - от легких (понос, сыпь) до тяжелых (печеночная недостаточность). Для лечения этого состояния назначаются соответствующие лекарства. Возникновение реакции "трансплантат против хозяина" не означает, что трансплантация прошла неудачно.
Аутологичная трансплантация
При этой процедуре донором стволовых клеток является сам пациент, находящийся в периоде ремиссии.
Кровь забирается из вены пациента на одной руке, проходит через прибор, отделяющий стволовые клетки, и возвращается в тело через вену другой руки.
Что такое рецидив при ХМЛ?
Слово рецидив используется, если была ремиссия. В отношении ХМЛ ремиссия может быть, во-первых, гематологической, то есть когда исчезают все внешние проявления заболевания (нормализация размеров печени и селезенки, исчезновение очагов поражения в органах), а также полная нормализация показателей периферической крови; во-вторых, цитогенетический, когда клетки с Филадельфийской хромосомой (Ph) уже не выявляются; и третий вариант, молекулярной, когда наиболее чувствительными молекулярно-генетическими методами (полимеразно-цепная реакция, ПЦР) продукт (транскрипт) патологического гена BCR-ABL не удается выявить. Существование молекулярной ремиссии спорно, так как возможность выявления транскрипта гена зависит от чувствительности используемого гена, качества реагентики и опыта сотрудников лаборатории. К тому же, чувствительность современных методик в целом ограничена. На сегодняшний день даже в наилучших лабораториях мира патологический транскрипт выявляется, если его количество больше, чем 1 на 100 000 транскрипта нормального контрольного гена. В вязи с этим слово молекулярная ремиссия в научной литературе заменена термином «негативность ПЦР».
Так как есть разные уровни ремиссии, то есть и разные уровни рецидива - гематологический (появление поражения разных органов, вновь ухудшение показателей клинического анализа крови), цитогенетический (появление Ph-позитивных клеток), молекулярный (вновь выявление транскрипта BCR-ABL).
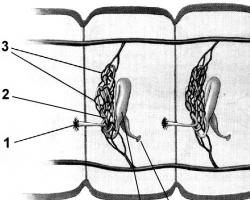
Аномальная Pb’-хромосома в клетке больного ХМЛ
Как можно обнаружить «рецидив ХМЛ»?
На сегодняшний день независимо от глубины достигнутой ремиссии, вплоть до молекулярной, рекомендуется постоянная непрерывная терапия ингибиторами тирозин киназ (ИТК). Перерывы в терапии или отмена препаратов показаны только из-за осложнений, связанных именно с ИТК. Естественно препараты также отменяют, если исходно не было эффекта или достигнутый эффект был позже утрачен.
На фоне терапии крайне важен мониторинг уровня лейкемических клеток с помощью не просто клинического анализа крови, но обязательно цитогенетического (особенно в течение первого года лечения) и ПЦР исследований. Именно молекулярно-генетические методики обнаруживают первые признаки рецидива болезни (появление лейкемических клеток) и свидетельствуют о возникшей неблагоприятной ситуации.
Почему возникает рецидив?
Причин множество и не все они изучены. Перечислю только наиболее яркие или изученные:
Как ни странно, нередкой причиной рецидива является неадекватный приём препарата пациентами. К сожалению, приходится сталкиваться с ситуацией, когда пациент самовольно снижает дозу препарата, вовсе перестаёт его принимать или принимает время от времени.
Одновременный длительный приём лекарств или веществ, снижающих концентрацию ИТК. Известно, что эти препараты разрушаются в печени под воздействием определенных ферментов - цитохромов. Есть большая группа лекарств или веществ, значительно повышающих активность этих цитохромов. В такой ситуации ИТК могут распадаться быстро, их концентрация резко снижается и, следовательно, снижается их эффективность. Поэтому, пациентов мы всегда предупреждаем о целесообразности сообщать нам, обо всех принимаемых препаратах. Мы не рекомендуем прием БАДов из-за невозможности оценить их влияние на концентрацию ИТК. К известным средствам, значительно активирующим цитохромы и снижающим активность ИТК относится, например, зверобой.
Эффективность ИТК также может снижаться из-за недостатка белков, которые «вкачивают» в клетку препараты или, наоборот, избытка белков, которые их «выкачивают» из клетки. Это может привести к снижению внутриклеточной концентрации ИТК.
Наиболее изученной причиной рецидивов следует считать появление мутаций (изменений) в гене BCR-ABL. Существует более 90 видов мутаций, которые могут менять структуру BCR-ABL белка. Не все они приводят к нарушению строения того участка белка, куда прикрепляются все ИТК. Поэтому, не все мутации одинаково плохо влияют на результаты лечения. К тому же для разных препаратов есть своя группа «плохих» мутаций. При этом есть одна мутация, появление которой приводит к неэффективности всех 3-х препаратов (иматиниб, нилотиниб, дазатиниб), зарегестрированных в России. Только ИТК под названием понатиниб (зарегистрирован в США в конце 2012 года) способен преодолеть те изменения, которые данная мутация привносит в клетку. Крайне важно выполнение мутационного анализа уже при появлении первых признаков неэффективности того или иного ИТК. Результаты данного анализа во многом помогают гематологу подобрать «правильный» ИТК для конкретного пациента.
Можно ли предотвратить рецидив?
Безусловно, можно, если причиной является неправильный приём препарата. Хотя не всегда удаётся исправить ситуацию впоследствии, но у части пациентов возобновление адекватной терапии приводит к улучшению ответа.
Риск рецидива также может быть снижен, если пациент не принимает препараты, влияющие на концентрацию ИТК.
Крайне важным является своевременное начало терапии ИТК сразу после постановки диагноза. Кроме того, в случае неэффективности первой линии ИТК, очень важна быстрая замена на другой ИТК. Всё это способствует быстрому снижению количества и активности лейкозных клеток. Это уменьшает риск развития в них дополнительных генетических изменений (мутации и др.), которые наиболее часто и приводят к рецидивам болезни даже после длительной ремиссии.
Учитывая все вышесказанное, наряду с соблюдением всех правил приёма препарата, крайне важно и своевременное обследование для оценки глубины ремиссии. Именно тщательный мониторинг (цитогенетический и/или ПЦР анализ) позволит врачу обнаружить рецидив болезни еще на ранних этапах и даст возможность своевременно назначить более эффективную терапию для повторного достижения ремиссии.
Хронический миелолейкоз в вопросах и ответах
Я заболел хмл. Что делать?
ХМЛ – хронический миелолейкоз, онкогематологическое заболевание крови, одна из разновидностей существующих лейкозов.
Хронический миелолейкоз – опухоль крови, не ограничивающая продолжительность жизни подавляющего большинства больных, благодаря современным препаратам и ответственному отношению к лечению.
Что делать? Довериться лечащему гематологу и соблюдать все рекомендации, а также своевременно проводить диагностические исследования.
Сколько я буду жить?
До недавнего времени средняя продолжительность жизни пациентов с ХМЛ составляла в среднем 3,5 года. Современные лекарственные средства позволяют продлить ее на срок более 20 лет, при этом качество жизни больных остается на высоком уровне и практически не отличается от жизни здорового человека.
Заразен ли хмл и передается ли он по наследству?
ХМЛ не является заразным заболеванием и не передается по наследству.
Может ли быть связан хмл с моей работой или экологией?
Влияние на заболеваемость ХМЛ таких факторов, как работа на вредном производстве, малые дозы радиации, слабое электромагнитное излучение, плохая экология мегаполисов и т.д., не нашли подтверждения. Поэтому не ищите причину, примите заболевание и научитесь с ним жить.
Почему лекарство нужно пить каждый день?
Лекарство необходимо принимать постоянно, пожизненно, без перерывов, т.к. в крови должна сохраняться определенная концентрация препарата. Самостоятельная отмена препарата может вызвать прогрессирование заболевания или препарат перестанет действовать на организм.
В какое время лучше принимать иматиниб?
Принимать препарат Иматиниб можно в удобное для вас время.
Последний прием Иматиниба должен быть не позднее, чем за 2 часа до сна.
Почему нельзя делать перерывы в приеме лекарства, когда перерывы необходимы?
Самостоятельная отмена препарата может вызвать потерю всех достигнутых результатов и привести к прогрессированию болезни (рецидиву). Перерывы в приеме препарата возможны только при наличии медицинских показаний и этот вопрос может решить только врач-гематолог.
Можно ли лечиться травами?
Не рекомендуется прием трав и БАДов из-за невозможности оценить их влияние на концентрацию препарата в крови. К известным средствам, значительно снижающим активность препарата, относятся, например, зверобой и женьшень.
Я слышал, что онкологию и хмл можно вылечить грибом чага или керосином?
К сожалению, вылечить народными средствами онкологию и ХМЛ невозможно.
Почему так часто необходимо сдавать анализы?
Анализы необходимы для контроля течения заболевания и своевременной корректировки лечения. Чтобы избежать повторного ухудшения самочувствия, каждое обследование необходимо проводить своевременно, когда назначил врач, и от этого зависит успех вашего лечения.
Почему недостаточно общего анализа крови?
Общий анализ крови - это забор крови из пальца или вены. Включает в себя подсчет лейкоцитов, эритроцитов, тромбоцитов и других компонентов крови, но этого анализа недостаточно, чтобы увидеть полную картину течения вашего заболевания.
Что такое цитогенетика? Обязательно ли ее сдавать?
Цитогенетический анализ – это забор костного мозга при стернальной пункции из грудины. При помощи этого исследования определяются хромосомные изменения и ответ на лечение, % клеток с филадельфийской хромосомой. Периодичность исследований определяет лечащий врач.
Что такое «молекулярная» диагностика?
Для проведения этого исследования сдают кровь из вены.
Молекулярный анализ является самым чувствительным методом диагностики ХМЛ из имеющихся.
Существует ли какая-то специальная диета при хмл?
Специальной диеты при заболевании ХМЛ не требуется.
Какие продукты нельзя принимать в пищу при хмл?
Можно ли пить витамины?
Необходимо быть крайне осторожными при ХМЛ с употреблением витаминов. Обязательно проконсультируйтесь с лечащим врачом.
Можно ли употреблять спиртное?
Не рекомендуется прием алкоголя, т.к. он может ускорять всасывание лекарств из пищеварительного тракта, создавая в организме более высокие концентрации препарата, чем при обычном приеме. Это приводит к передозировке или развитию токсических реакций, которые пагубно влияют на печень, как и сам алкоголь.
Можно ли мне работать?
В целом ХМЛ не влияет на вашу работоспособность, однако необходимо помнить о соблюдении режима лечения.
Можно ли мне заниматься спортом?
Посоветуйтесь со своим лечащим врачом.
Можно ли ходить в баню?
Можно ли отдыхать на море?
Отдыхать можно и нужно, с соблюдением простых правил:
Закрытая одежда и панамка;
Использование зонта (тента);
Не перегреваться.
Однако,
Можно ли загорать?
Не противопоказано пребывание на солнце до 11 утра и после 5 вечера с применением солнцезащитных кремов.
У меня сильная «побочка». Что делать?
Лечение ХМЛ часто сопровождается побочными эффектами, которые зависят от дозы препарата, фазы ХМЛ, длительности лечения, пола, возраста. Реакция разных людей на препараты индивидуальна, поэтому ваши побочные реакции могут отличаться от реакции других пациентов. При возникновении побочных эффектов, не прекращайте прием препарата, немедленно проконсультируйтесь со своим врачом, так как некоторые побочные эффекты требуют лечения.
Тошнота
Попробовать подобрать или исключить во время приема лекарства какие-то определенные продукты. Например, можно заесть зеленым яблоком. Исключить молочные, кислые и копченые продукты.
Изжога
Ограничить переедание, острые приправы, кофеин и алкоголь.
Не ложиться спать в течение 1-2 часов после приема иматиниба.
Задержка жидкости с развитием отеков
Ограничить прием соли в рационе, уменьшить объем употребляемой жидкости (особенно на ночь).
Косметические процедуры.
Необходима консультация у лечащего врача.
Диарея (понос)
Постарайтесь исключить такие продукты как чернослив, свекла, молочные и т.д.
Необходима консультация у лечащего врача.
Курага, фасоль, бобовые, крупы, мясо, морская капуста, свежие шампиньоны, картофель (особенно запеченный или сваренный в мундире), морковь, свекла, тыква, редька, перец, помидоры, огурцы, капуста, зелень (особенно шпинат и петрушка);
Яблоки, бананы, арбузы, дыни, киви, манго, авокадо, вишня, виноград, черная смородина, крыжовник, ежевика, сухофрукты (инжир, курага, чернослив, финики), орехи (особенно грецкие и фундук).
Кешью, гречневая крупа, гречиха, пшено, отруби, бобовые (особенно белая фасоль и соя), морковь, картофель, шпинат и другие листовые овощи абрикосы, персики, бананы, ежевика, малина, клубника, кунжут, орехи.
Кожные высыпания
Необходима консультация лечащего врача.
Повышение температуры, лихорадка
Такая реакция возможна на действие препарата.
Необходима консультация лечащего врача.
Я заболел (простудой, гриппом и т.д.). Что делать?
Не занимайтесь самолечением, проконсультируйтесь с лечащим врачом.
Мне назначены лекарственные препараты по сопутствующему заболеванию, можно ли их принимать вместе с иматинибом?
Проконсультируйтесь со специалистами.
В случае появления жалоб на фоне лечения необходимо обратиться к лечащему врачу для решения вопроса о необходимости корректировки терапии.
Нужна ли инвалидность для получения лекарственных препаратов?
Для получения лекарственных препаратов для лечения ХМЛ не требуется получение инвалидности.
Онкологическим больным все лекарственные средства положены бесплатно на основании действующего законодательства.
В каждом случае оформление инвалидности вопрос индивидуальный и вам самим необходимо определить для себя, нужна инвалидность или нет. А присвоят инвалидность или нет, будет зависеть от медицинских показаний.
Дают ли группу инвалидности при хмл?
Да, дают. Для получения группы инвалидности должны быть медицинские и социальные показания.
Критерии для МСЭ: Заболевание имеет неблагоприятный прогноз. Появление признаков акселерации, развитие бластного криза, свидетельствующие о тяжелых нарушениях функций и плохом прогнозе.
Критерии инвалидности.
III группа инвалидности определяется больным, у которых диагностирована хроническая фаза, при достижении клинико-гематологической ремиссии, адекватного снижения лейкоцитоза, при наличии ограничения способности к трудовой деятельности I ст., что требует рационального трудоустройства в не противопоказанных условиях и видах труда или уменьшения объема выполняемой работы.
II группа инвалидности определяется больным с прогрессированием заболевания, при отсутствии полной клинико-гематологической ремиссии и адекватного снижения лейкоцитоза; развитии осложнений, ограничении способности к самообслуживанию, передвижению и трудовой деятельности II ст.
I группа инвалидности определяется больным при наличии бластного криза, фазы акселерации, тяжелых гнойно-септических осложнений, ограничение способности к самообслуживанию и передвижению III ст. Больные нуждаются в постоянной посторонней помощи.
Мы хотим ребенка
Планирование беременности и для здоровых людей непростой вопрос, а для пациентов с ХМЛ этот вопрос требует ответственного решения. Но в любом случае, это решение должно приниматься только при участии лечащего врача, т.к. потребует корректировки лечения.
На сегодняшний день у пациентов с ХМЛ рождаются здоровые дети практически во всех регионах страны.
Что такое препараты 2-ой линии?
На сегодняшний день имеется ряд лекарственных препаратов, назначаемых пациентам, которым не помогает Иматиниб. Они гораздо сильнее по своему действию, но подбор для каждого пациента осуществляется индивидуально.
Как взаимодействует иматиниб с другими лекарственными препаратами?
При назначении лечения сопутствующих заболеваний другими специалистами необходимо заключение Вашего врача гематолога.
Необходимо обратить внимание:
Индукторы CYP3A4/5 - препараты, снижающие концентрацию ИТК в плазме:
Глюкокортикоиды, гризеофульвин, дексаметазон, дифенин, карбамазепин, окскарбазепин, прогестерон, рифабутин, рифампицин, сульфадимизин, троглитазон, фенилбутазон, фенобарбитал, этосуксимид.
Ингибиторы CYP3A4/5 - препараты, повышающие концентрацию ИТК в плазме:
Азитромицин, амиодарон, анастрозол, верапамил, гестоден, грейпфрутовый сок, даназол, дексаметазон, дилтиазем, диритромицин, дисульфирам, зафирлукаст, изониазид, итраконазол, метронидазол, мибефрадил, миконазол (средний), норфлоксацин, оксиконазол, омепразол (слабый), пароксетин (слабый), сертиндол, сертралин, флувоксамин, флуоксетин, хинидин, хинин, циклоспорин, кетоконазол, циметидин, кларитромицин, эритромицин, клотримазол, этинилэстрадиол
Препараты, удлиняющие интервал QT
- Антиаритмические: аденозин, амиодарон, флекаинид, хинидин, соталол.
- Противосудорожные: фелбамат, фенитоин.
- Антидепрессанты: амитриптилин, циталопрам, дезипрамин, доксепин, имипрамин, па роксетин, сертралин.
- Антигистаминные: астемизол, дифенгидрамин, лоратадин, терфенадин.
- Антигипертензивные: индапамид, мибефрадил, гидрохлортиазид, нифедипин.
- Противомикробные: макролиды, фторхинолоны.
- Противоопухолевые: триоксид мышьяка, тамоксифен.
- Нейролептики: хлорпромазин, клозапин, дроперидол, галоперидол, рисперидон.
- Препараты, действующие на желудочно-кишечный тракт: цизаприд, доласетрон, октреотид.
- Препараты разных групп: амантадин, метадон, салметерол, суматриптан, такролимус.