Синдром Франческетти Описан в 1944 году женевским профессором A. Franceschetti и его учеником Zwahlen и назван «мандибуло-фациальный дизостоз». В литературе иногда он описывается под названием «синдром Franceschetti-Zwahlen». Обусловлен синдром дисплазией эмбрионального элемента первой жаберной дуги неизвестного происхождения (Кручинский Г.В., 1972)

Наследственность Болезнь имеет семейно-наследственный характер и передается по аутосомно-доминантному типу. Наблюдается у членов одной и той же семьи и даже в двух и трех генерациях. Для заболевания характерна высокая пенетрантность (т.е. высокая вероятность проявления признаков болезни у людей с мутацией) и различная экспрессивность (т.е. различный характер и тяжесть проявления болезни). Молекулярно-генетической причиной заболевания являются, как правило, нонсенс мутации в гене TCOF1, приводящие к возникновению преждевременного стоп кодона и, как следствие, к гаплонедостаточности (состояние, при котором половинного количества генного продукта недостаточно для нормального функционирования организма).

Клинические проявления Синдром характеризуется разнообразием челюстно-лицевых аномалий в различных комбинациях. При полной форме синдрома со стороны глаз отмечаются: косые «анти монголоидные» глазные щели, т. е. двустороннее опущение наружного угла глазной щели. На нижнем, реже верхнем веке снаружи колобома века, отсутствие мейбомиевых желез. Наблюдаются эпибульбарные дермоиды, парез глазодвигательных мышц, редко микрофтальм, врожденная катаракта, колобома сосудистого тракта и зрительного нерва.

Клинические проявления Со стороны челюстно-лицевой системы имеется гипоплазия костей лица, скуловые кости мелкие и недоразвитые, что приводит к значительной асимметрии лица. Скуловые отростки височных костей могут отсутствовать, тело и ветви челюстных костей иногда недоразвиты. Наблюдаются гипоплазия нижней челюсти, прогнатизм, макростомии, высокое небо, значительно реже его расщепление. Недоразвитая нижняя челюсть придает лицу птичий вид.

Клинические проявления Из зубных аномалий имеется резкое недоразвитие зубов, в первую очередь моляров, зубы широко расставлены, нередко имеется аномалия прикуса. Часто наблюдаются врожденные уродства ушей, их аплазия, иногда «ложные уши», свищи между углом рта и ухом, гиперплазия лобных пазух и недоразвитие гайморовых. Наблюдается также атипичный рост волос низко на лбу, на щеках, иногда частичная алопеция. Изредка отмечаются увеличение языка, отсутствие околоушной железы, внутренняя гидроцефалия, поражение сердца и крупных сосудов, трахеи, бронхов, крипторхизм, умственное недоразвитие.

Дифференциальная диагностика Дифференцируют с синдромом глазо-ушно-позвоночной дисплазии Гольденхаара, при котором имеются эпибульбарные дермоиды, часто связанные с колобомой верхнего века, ушные отростки и предушные слюнные фистулы, нарушение строения наружного слухового прохода и тугоухость. Кроме того, имеются аномалии позвоночника, шейный синостоз, увеличение количества грудных или поясничных позвонков и др. Часто наблюдается микростомия и небольшое уменьшение лица, высокое расщепленное небо, раздвоенный язык и аномалии зубов. Некоторые авторы объединяют эти 2 синдрома в один и называют его синдромом Франческетти-Гольденхаара.

Дифференциальная диагностика Возникновение обоих синдромов связано с одним и тем же периодом внутриутробного развития, на 7-9-й неделе развития плода. Но синдром Франческетти является наследственным. Эти же авторы подробно изучали эмбриологическое развитие орбиты, неба и нижней челюсти и объясняли частое совпадение врожденных аномалий глаз, его придатков с изменениями неба и нижней челюсти общностью происхождения их из дериватов первой жаберной дуги.

Лечение Несмотря на наличие разнообразных аномалий, прогноз при синдроме Франческетти благоприятный. Этиологического лечения не существует, проводят по возможности пластические операции. В лечении пациентов, пострадавших от ТКС, применяется междисциплинарный подход, то есть требуются вмешательства различных специалистов. Основные проблемы у пациентов с ТКС нарушения глотания и проходимости дыхательных путей. Некоторые пациенты нуждаются в трахеостомии. Гастростома может быть необходима для обеспечения адекватного потребления пищи и для защиты дыхательных путей. Вопрос о хирургическом восстановлении структуры лица решается индивидуально и осуществляется по достижении определённого возраста.

Лечение Дефекты ушной раковины также устраняются хирургическим способом. Но даже в тех случаях, когда ушные раковины и наружные слуховые проходы не затронуты синдромом, цепи слуховых косточек часто повреждены. Поэтому также необходима слуховая реабилитация с фиксируемыми к кости слуховыми аппаратами (BAHAs) и другие способы на основе костной проводимости. Челюстно-лицевая и пластическая хирургия позволяет исправить гипоплазию мягких тканей, гипоплазию костей (вытяжение, костные трансплантаты), колобому века и волчью пасть.


Ключевые слова
АНОМАЛИЯ РАЗВИТИЯ / ГЕННЫЕ МУТАЦИИ / GENE MUTATIONS / ГОЛОВНАЯ БОЛЬ / HEADACHE / СИНДРОМ ФРАНЧЕСКЕТТИ / FRANCESCHETTI SYNDROME / DEVELOPMENTAL ANOMALYОписывается клинический опыт ведения пациента с врожденным заболеванием (мандибуло-фациальным дизостозом), возникающим в результате поражения структур, исходящих из первой жаберной дуги. Это патологическое состояние наследуется по аутосомно-доминантному типу. Заболевание может наблюдаться в двух или даже трех генерациях. Данное наблюдение представляет большой интерес с клинической точки зрения, поскольку крайне редко встречается в повседневной практике. Ранняя диагностика данного генетического синдрома представляет большие сложности. По мнению авторов статьи, в подобных ситуациях оправдана постановка синдромо-логического диагноза с уточнением аномалий развития на основании анализа совокупности клинических данных, дополнительных методов обследования, с последующей хирургической коррекцией нарушенных функций.
Головная боль в практике педиатра: многофакторный анализ клинико-социальных предикторов
2015 / Ахмадеева Л.Р., Валеева Д.С., Вейцман Б.А., Ахмадеева Э.Н.Ахондрогенез второго типа (синдром пангера - Салдино) у новорожденного: описание клинического случая
2015 / Черненков Ю.В., Нечаев В.Н., Стасова Ю.В., Терещенко В.А.Показатели заболеваемости ретинопатией недоношенных детей в учреждениях родовспоможения Саратовской области
2015 / Черненков Ю.В., Нечаев В.Н., Терещенко В.А., Стасова Ю.В.Клиническое наблюдение врожденной альвеолярно-капиллярной дисплазии легких у новорожденного ребенка
2016 / Гаврилова Е.С., Борисова А.А., Быкова Е.В., Позгалёва Н.В., Панина О.С., Черненков Ю.В.Оценка показателей здоровья детей, рожденных с помощью применения репродуктивных технологий
2014 / Черненков Ю. В., Нечаев В. Н., Стасова Ю. В.Редкий клинический случай в неонатальной практике: врожденный порок развития стенки желудка с ее спонтанным разрывом
2015 / Черненков Ю.В., Лаврова Д.Б., Панина О.С, Прокопенко Л.Е, Ларшина Е.П., Шиханова С.В., Беляева Н.А.Клиническое наблюдение: врождённая кистозная мальформация легкого
2014 / Черненков Ю. В., Горемыкин И. В., Бочкова Л. Г., Клюев С. А.Клинический случай наследственной гидроцефалии (синдром Денди-Уокера)
2016 / Черненков Ю.В., Нечаев В.Н., Лысова Ю.В.Оценка валеологических знаний девочек-подростков, учащихся средних школ и учреждений начального профессионального образования
2013 / Кунина А. В., Кунина С. В., Гуменюк О. И., Черненков Ю. В.The aim of the article is to present the clinical experience of conducting the patient with a congenital disease (mandibulo-facial dysostosis) resulting from defeat of the structures proceeding from the first branchial arch. The pathological state is inherited on autosomno-dominanttype. The disease can be observed in two or even three generations. The supervision represents a great interest from the clinical point of view as it rarely occurs in daily practice. Early diagnostics of difficult genetic syndromes and the clinical supervision described represent significant difficulties. According to the research, in similar situations statement of the syndromal diagnosis with specification of anomalies of development on the basis of the analysis of clinical data, additional methods of inspection, with the subsequent surgical correction of the broken functions has been justified.
отношение к биологическим и социальным характеристикам, и основывалась как на объективных, так и на субъективных показателях, которые были получены при осмотре и опросе как самих детей, так и их родителей. Для проверки эффективности модели на других популяциях требуется дополнительное исследование с включением большего количества пациентов с головными болями напряжения детского возраста.
Заключение. Знание предикторов ГБ в целом, а также факторов её хронизации позволяет на этапе диагностики ГБ выделить факторы неблагоприятного течения ГБ, в том числе факторы, способствующие увеличению частоты приступов. Работа с пациентом, страдающим ГБ, и особенно с ребенком, требует индивидуального подхода, тщательного сбора жалоб, в том числе путем активного опроса. Разработанная нами модель проста и может быть предложена в практической медицине для использования при составлении предварительного прогноза для детей с головными болями напряжения, а выделенные параметры предлагаем учитывать при беседе с пациентами и их семьями и профилактической работе.
Конфликт интересов. Мы благодарим TeX Users Group за финансовую поддержку участия одного из авторов (Л. Р. А.) в конференции, во время которой мы (Л. Р. А и Б. А. В.) смогли обсудить данную работу и продолжить подготовку статьи.
References (Литература)
1. Straube A, Heinen F, Ebinger F, et al. Headache in School Children: Prevalence and Risk Factors. Deutsches arzteblatt international 2Q13; 1Ю (48): 811-818.
2. Urazbagambetov A, Delyagin VM. Headaches in infants and adolescents. Practical medicine 2Q14; 2 (78): 42-44. Russian
(Уразбагамбетов А., Делягин В. М. Головные боли у детей и подростков. Практическая медицина 2014; 2 (78): 42-44.)
3. Valeeva DS, Akhmadeeva EN, Akhmadeeva LR. Influence of lifestyle factors on frequency of headaches in children. Rossiyskiy zhurnal boli 2014; 1 (42): 89. Russian (Валеева Д. С., Ахмадеева Э. Н., Ахмадеева Л. Р. Влияние факторов образа жизни на частоту головных болей у детей. Российский журнал боли 2014; 1 (42): 89.)
4. Izmaylova IG, Belopasov VV, Dzhumagaziev AA, et al. Forecasting of chronic tension-type headache in children and adolescents. Psikhicheskoe zdorov"e 2014; 12 (7): 41-48. Russian (Измайлова И. Г., Белопасов В. В., Джумагазиев А. А. и др. Прогнозирование хронического течения головной боли напряжения у детей и подростков. Психическое здоровье 2014; 12 (7): 41-48.)
5. Izmaylova IG. Comorbid disorders in children and adolescents with primary headaches. Psikhicheskoe zdorov"e 2012; 10 (6): 37-43. Russian (Измайлова И. Г. Коморбидные расстройства у детей и подростков с первичными цефалги-ями. Психическое здоровье 2012; 10 (6): 37-43.)
6. Sergeev AV, Rachin AP, Avdeeva TG. Tension-type headache in children with thyroid gland and gastro-intestinal diseases. Rossiyskiy zhurnal boli 2013; 2 (39): 13-18. Russian (Сергеев А. В., Рачин А. П., Авдеева Т. Г. Головная боль напряжения у детей с патологией щитовидной железы и желудочно-кишечного тракта. Российский журнал боли 2013; 2 (39): 13-18.)
7. Headache Classification Committee of the International Headache Society. The International Classification of Headache Disorders, 3rd edition. Cephalalgia 2013; 33 (9): 629-808.
8. R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria (2015). http://www.R-project.org/ (11 July 2015).
9. Venables WN, Ripley BD. Modern Applied Statistics with S. 4th ed. New York: Springer-Verlag, 2002; 498 p.
10. Sing T, Sander O, Beerenwinkel N, et al. ROCR: visualizing classifier performance in R. Bioinformatics 2005; 21 (20): 3940-3941. http://rocr.bioinf.mpi-sb.mpg.de> (11 July 2015).
УДК 616.716-007.17-008.6-053.31-07-08 (045) Клинический случай
СИНДРОМ ФРАНЧЕСКЕТТИ (МАНДИБУЛО-ФАЦИАЛЬНЫЙ ДИЗОСТОЗ) У НОВОРОЖДЕННОГО: КЛИНИЧЕСКИЙ СЛУЧАЙ
В. Н. Нечаев - ГБОУ ВПО «Саратовский ГМУ им. В. И. Разумовского» Минздрава России, доцент кафедры госпитальной педиатрии и неонатологии, кандидат медицинских наук; В.А. Терещенко - ГБОУ ВПО «(Саратовский ГМУ им. В. И. Разумовского» Минздрава России, ординатор кафедры госпитальной педиатрии и неонатологии; Ю. В. Стасова - ГБОУ ВПО «(Саратовский ГМУ им. В. И. Разумовского» Минздрава России, ординатор кафедры госпитальной педиатрии и неонатологии; Ю. В. Черненков - ГБОУ ВПО «(Саратовский ГМУ им. В. И. Разумовского» Минздрава России, проректор по науке, заведующий кафедрой госпитальной педиатрии и неонатологии, профессор, доктор медицинских наук.
FRANCESCHETTI SYNDROME (MANDIBULO-FACIAL DYSOSTOSIS) AT A NEWBORN: CLINICAL CASE
V. N. Nechaev - Saratov State Medical University n.a. V. I. Razumovsky, Department of Hospital Pediatrics and Neonatology, Assistant Professor, Candidate of Medical Science, V. A. Tereshenko - Saratov State Medical University n.a. V. I. Razumovsky, Department of Hospital Pediatrics and Neonatology, Post-graduate; Yu. V. Stasova - Saratov State Medical University n.a. V. I. Razumovsky, Department of Hospital Pediatrics and Neonatology, Post-graduate; Yu. V. Chernenkov - Saratov State Medical University n.a. V. I. Razumovsky, Head of Department of Hospital Pediatrics and Neonatology, Professor, Doctor of Medical Science;
Дата поступления - 3.07.2015 г Дата принятия в печать - 10.12.2015 г.
Нечаев В. Н., Терещенко В.А., Стасова Ю. В., Черненков Ю. В. Синдром Франческетти (мандибуло-фациальный дизостоз) у новорожденного: клинический случай. Саратовский научно-медицинский журнал 2015; 11 (4): 551-553.
Описывается клинический опыт ведения пациента с врожденным заболеванием (мандибуло-фациальным дизостозом), возникающим в результате поражения структур, исходящих из первой жаберной дуги. Это патологическое состояние наследуется по аутосомно-доминантному типу. Заболевание может наблюдаться в двух или даже трех генерациях. Данное наблюдение представляет большой интерес с клинической точки зрения, поскольку крайне редко встречается в повседневной практике. Ранняя диагностика данного генетического синдрома представляет большие сложности. По мнению авторов статьи, в подобных ситуациях оправдана постановка синдромо-логического диагноза с уточнением аномалий развития на основании анализа совокупности клинических данных, дополнительных методов обследования, с последующей хирургической коррекцией нарушенных функций.
Ключевые слова: генные мутации, аномалия развития, синдром Франческетти.
Saratov Journal of Medical Scientific Research. 2Q15. Vol. 11, № 4.
ПЕДИАТРИЯ
Nechaev VN, Tereshenko VA, Stasova YuV, Chernenkov YuV. Franceschetti syndrome (mandibulofacial dysostosis) at a newborn: Saratov Journal of Medical Scientific Research 2015; 11 (4): 551-553.
The aim of the article is to present the clinical experience of conducting the patient with a congenital disease (mandibulofacial dysostosis) resulting from defeat of the structures proceeding from the first branchial arch. The pathological state is inherited on autosomno-dominant type. The disease can be observed in two or even three generations. The supervision represents a great interest from the clinical point of view as it rarely occurs in daily practice. Early diagnostics of difficult genetic syndromes and the clinical supervision described represent significant difficulties. According to the research, in similar situations statement of the syndromal diagnosis with specification of anomalies of development on the basis of the analysis of clinical data, additional methods of inspection, with the subsequent surgical correction of the broken functions has been justified.
Key words: gene mutations, developmental anomaly, Franceschetti syndrome.
Введение. Мандибуло-фациальный дизостоз - это поражение структур, исходящих из первой жаберной дуги, наследуемый по аутосомно-доминант-ному типу с высокой (до 90%) пенетрантностью и различной экспрессивностью переменной, даже среди больных одной семьи. Может наблюдаться в двух или даже трех генерациях . Этот синдром вызывается мутациями в TCOF1 гене (5q32-q33.1) или в POLR1C гене (6p21.1) и POLR1D гене (13q12.2), кодирующих РНК-полимеразы I и III подразделений . Он обусловлен дисплазией эмбрионального элемента первой жаберной дуги неизвестного происхождения . Описывается как врожденное нарушение развития костей черепа (чаще височной кости) и лица, характеризуется двусторонней симметричной отонижнечелюстной дисплазией без аномалий развития конечностей, он также связан с некоторыми дефектами головы и шеи .
Синдом Франческетти - редкое заболевание, встречающееся в одном случае на 50000 детей, рожденных живыми. Дифференциальная диагностика проводится с синдромом глазоушно-позвоноч-ной дисплазии Гольденхаара, при которой имеются эпибульбарные дермоиды, часто связанные с коло-бомой верхнего века, ушные отростки и предушные слюнные фистулы, нарушение строения наружного слухового прохода и тугоухость, шейный синостоз, увеличение количества грудных или поясничных позвонков и другие аномалии позвоночника. Часто наблюдается высокое расщепленное небо, раздвоенный язык и аномалии зубов .
Несмотря на наличие разнообразных аномалий, прогноз при синдроме Франческетти благоприятный. Этиологического лечения не существует, по возможности проводят пластические операции. Гибель пациентов чаще наступает от интеркуррентных заболеваний.
Описание клинического случая. Под нашим наблюдением находилась новорожденная доношенная девочка Мария Г., родившаяся от 6-й беременности, протекавшей на фоне отеков, вызванных беременностью, хронической внутриутробной гипоксии плода, фетоплацентарной недостаточности, вегетососуди-стой дистонии по гипертоническому типу, хронического гастрита. В 20 недель мать проходила лечение по поводу острого токсоплазмоза, отмечалось много-водие, в 34 недели у ребенка выявлен порок мочевы-водящей системы (МВС). Роды третьи срочные, в головном предлежании. Масса ребенка при рождении 2800 гр, рост 50 см, с оценкой по шкале Апгар 4,5; 5 баллов.
Из родильного зала ребенок поступил в ОРИТН в тяжелом состоянии за счет явлений дыхательной недостаточности 3 степени, неврологической сим-
Тел.: 89053296726
E-mail: [email protected]
птоматики, задержки внутриутробного развития, кожного геморрагического синдрома. С рождения до 20-х суток жизни проводилась респираторная терапия, затем в течение 7 дней дыхание через воздуховод, в последующем спонтанное, лишь при нарастании дыхательной недостаточности периодически нуждалась в постановке воздуховода. Проводилось зондовое кормление.
Обращали на себя внимание множественные пороки развития: выраженная микрогения, гипоплазия скуловых костей, большой «клювовидный нос», короткий фильтр, глазной гипотеларизм, микрофталь-мия, антимонголоидный разрез глаз, «птичье лицо», деформация ушных раковин (низко расположенные, большие уши), гипоплазия ногтевых пластинок (на кистях и стопах), широкая грудная клетка, сосковый гипертеларизм, выраженная бледность кожных покровов. В связи с выявлением пороков развития и более 5 стигм дисэмбриогенеза ребенок консультирован генетиком. На основании фенотипических данных обследования методом синдромологическо-го анализа был заподозрен синдром Франческетти: аутосомно-доминантное заболевание.
Пациентке было проведено кариотипирование, выявлен нормальный женский кариотип (46 хх).
Ребенок находился на зондовом питании, на третьи сутки жизни девочка была проконсультирована детским хирургом по поводу непроходимости носовых ходов. Поставлен диагноз: «ВПР Атрезия хоан».
Кроме того, дополнительно потребовался осмотр отоларинголога, проведена попытка зондирования носовых ходов зондом №4. Диагноз: «Атрезия хоан слева, сужение носового хода справа». Было рекомендовано хирургическое лечение в плановом порядке.
Отоакустическая эмиссия у ребенка не была зарегистрирована (заподозрена тугоухость).
На 23-и сутки жизни, после стабилизации состояния, девочка была переведена в отделение патологии новорожденных и недоношенных детей для дальнейшего обследования и лечения.
По данным нейросонографии выявлена гипоплазия мозолистого тела, расширение межполушарной щели и субарахноидального пространства, дилата-ция 3-го желудочка мозга.
После этого девочка была осмотрена неврологом и выставлен диагноз: «ВПР головного мозга: Микроцефалия? Гипоплазия мозолистого тела. Последствия гипоксического поражения нервной системы, синдром двигательных нарушений. Задержка психомоторного развития, ранний восстановительный период».
По результатам ДЭХО-КГ выявлена коарктация аорты, функционирующее овальное окно до 0,47 см, открытый артериальный проток - 0,4 см. Диагноз подтвержден кардиохирургом, было рекомендовано оперативное лечение в более позднем периоде.
Саратовский научно-медицинский журнал. 2015. Т. 11, № 4.
После осмотра окулистом у девочки выявлена микрофтальмия, а также атрофия дисков зрительных нервов обоих глаз.
По данным УЗИ брюшной полости обнаружена тазовая дистопия и гипоплазия левой почки. С четвертой недели жизни появились изменения в моче в виде лейкоцитурии до 25 в поле зрения, в динамике уровень лейкоцитов нарос до 40, появилась протеинурия. В посеве мочи на стерильность выявлен умеренный рост E. Coli, была назначена антибактериальная терапия. Девочка осмотрена детским урологом, и, учитывая наличие врожденного порока развития МВС, заподозрено наличие пузырно-моче-точникового рефлюкса в гипоплазированную почку. Было рекомендовано: УЗИ до и после микции, продолжить проведение антибактериальной терапии, поставить постоянный катетер, контроль ОАМ один раз в 2 недели, а также наблюдение и обследование ребенка урологом в плановом порядке, после коррекции аномалий развития.
Перед выпиской из отделения ребенок повторно осмотрен генетиком и, с учетом выявленных множественных пороков развития (атрезия хоан слева и сужение справа, сформировавшаяся микроцефалия, гипоплазия мозолистого тела, тазовая дистопия и гипоплазия левой почки, ВПС - коарктация аорты, ОАП, а также множественные стигмы дисэмбриоге-неза), синдром Франческетти был подтвержден.
За время нахождения в стационаре девочке проводилось симптоматическое лечение, направленное на поддержание витальных функций и усиление адаптационных возможностей организма, антибактериальная, ноотропная, инфузионная терапия и парентеральное питание.
После проведенного лечения и объяснения маме особенностей ухода за ребенком, необходимости хирургической лечения по восстановлению проходимости носовых ходов, коррекции ВПС, необходимости
динамического наблюдения узких специалистов ребенок переведен ОДКБ.
Заключение. Настоящее наблюдение представляет большой интерес с клинической точки зрения, поскольку заболевание крайне редко встречается в повседневной практике. Ранняя диагностика сложных генетических синдромов, к которым относится описываемое нами клиническое наблюдение, представляет большие сложности. По нашему мнению, в подобных ситуациях оправдана постановка син-дромологического диагноза с уточнением аномалий развития на основании анализа совокупности клинических и лабораторных данных, дополнительных методов обследования, с последующей хирургической коррекцией выявленных нарушений и проведением реабилитационных мероприятий.
Конфликт интересов не заявляется.
References (Литература)
1. Kozlova SI, Demikova NS, Semanova E, Blinnikova OE. Hereditary syndromes and medico-genetic consultation. Moscow: Practice, 1996; р. 122-189. Russian (Козлова С. И., Демикова Н. С., Семанова Е, Блинникова О. Е. Наследственные синдромы и медико-генетическое консультирование. М: Практика, 1996; с. 122-189).
2. Crow YZ, et al. Syndrome displays genetic heterogeneity with one locus (5q32-q33.1), POLR1C (6p21.1) and POLR1D (13q12.2). Am J Hum Genet 2000; 67 (1): 213-221.
3. Lazyuk GI, Kruchinsky GV, Kirillov IA. Teratologiya of the person. Moscow: Practice, 1991; р. 27-152. Russian (Ла-зюк Г. И., Кручинский Г. В, Кириллова И. А. Тератология человека. М: Практика, 1991; с. 27-152).
4. Loreena LV. Questions of medical genetics. Ryazan: RyazSMU, 2011; р. 8-21. Russian (Лорина Л. В. Вопросы медицинской генетики. Рязань: РязГМу, 2011; с. 8-21).
5. Mutovin GR. Fundamentals of clinical genetics. Moscow: The Higher School, 2001; р. 49-53. Russian (Мутовин Г. Р. Основы клинической генетики. М.: Высшая школа, 2001; с. 4953).
6. Schwartz UG. The chosen questions of clinical genetics. Saratov: SSMU, 2011; 88 p. Russian (Шварц Ю. Г. Избранные вопросы клинической генетики. Саратов: СГМУ, 2011; 88 с.).
УДК617.735-053.32:314.44 (470.44) »45=02»(045) Оригинальная статья
ПОКАЗАТЕЛИ ЗАБОЛЕВАЕМОСТИ РЕТИНОПАТИЕЙ НЕДОНОШЕННЫХ ДЕТЕЙ В УЧРЕЖДЕНИЯХ РОДОВСПОМОЖЕНИЯ САРАТОВСКОЙ ОБЛАСТИ
Ю. В. Черненков - ГБОУ ВПО «Саратовский ГМУ им. В. И. Разумовского» Минздрава России, проректор по науке, заведующий кафедрой госпитальной педиатрии и неонатологии, профессор, доктор медицинских наук; В. Н. Нечаев- ГБОУ ВПО «Саратовский ГМУ им. В. И. Разумовского» Минздрава России, доцент кафедры госпитальной педиатрии и неонатологии, кандидат медицинских наук; В. А. Терещенко - ГБОУ ВПО «Саратовский ГМУ им. В. И. Разумовского» Минздрава России, ординатор кафедры госпитальной педиатрии и неонатологии; Ю. В. Стасова - ГБОУ вПо «Саратовский ГМУ им. В. И. Разумовского» Минздрава России, ординатор кафедры госпитальной педиатрии и неонатологии.
INDICES OF RETINOPATHY MORBIDITY AMONG PREMATURE CHILDREN IN SARATOV REGION INSTITUTIONS OF OBSTETRICS
Yu. V. Chernenkov - Saratov State Medical University n.a. V. I. Razumovsky, Head of Department of HospitalPediatrics and Neonatology, Professor, Doctor of Medical Science; V. N. Nechaev - Saratov State Medical University n.a. V. I. Razumovsky, Department of HospitalPediatrics and Neonatology, Professor Assistant, Candidate of Medical Science;V. A. Tereshenko - Saratov State Medical University n.a. V. I. Razumovsky, Department of HospitalPediatrics and Neonatology, Post-graduate; Yu. V. Stasova - Saratov State Medical University n.a. V. I. Razumovsky, Department of HospitalPediatrics and Neonatology, Post-graduate.
Дата поступления -3.07.2015 г. Дата принятия в печать - 10.12.2015 г.
Черненков Ю.В., Нечаев В.Н., Терещенко В.А., Стасова Ю.В. Показатели заболеваемости ретинопатией недоношенных детей в учреждениях родовспоможения Саратовской области. Саратовский научно-медицинский журнал 2015; 11 (4): 553-555.
Цель: изучить эффективность диагностических и профилактических мероприятий по снижению частоты и тяжести ретинопатии недоношенных (РН), а также провести анализ заболеваемости, сопутствующих состояний и динамический мониторинг недоношенных детей с РН. Материали методы. За 2 года был обследован на
Saratov Journal of Medical Scientific Research. 2015. Vol. 11, № 4.
При внутриутробных нарушениях процессов развития костей возникают серьезные черепно-лицевые деформации, и одной из разновидностей такой патологии является синдром Тричер Коллинза (TCS) или мандибулофасциальный, то есть челюстно-лицевой дизостоз.
Код заболевания по МКБ 10: класс XVII (врожденные аномалии, деформации и хромосомные нарушения), Q75.4 - mandibulofacial dysostosis.
Q75.4 Челюстно-лицевой дистоз
Данный синдром получил имя выдающегося британского офтальмолога Эдварда Тричер Коллинза, описавшего основные черты патологии более ста лет назад. Однако европейские врачи чаще называют этот вид аномалии костей лица и челюстей болезнью или синдромом Франческетти – на основании обширных исследований швейцарского офтальмолога Адольфа Франческетти, который ввел термин «мандибулофасциальный дизостоз» в середине прошлого века. В медицинских кругах также используется название – синдром Франческетти-Коллинза.
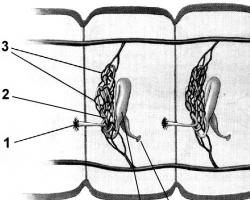
Причины синдрома Тричер Коллинза – мутации гена TCOF1 (в локусе хромосомы 5q31.3-33.3), который кодирует ядрышковый фосфопротеин, отвечающий за формирование черепно-лицевой части эмбриона человека. В результате преждевременного уменьшения количества этого белка нарушаются биогенез и функции рРНК. По мнению генетиков исследовательской программы Human Genome, эти процессы приводят к сокращению пролиферации эмбриональных клеток нервного гребня – валика вдоль нервного желоба, который в ходе развития зародыша замыкается в нервную трубку.
Формирование тканей лицевой части черепа происходит благодаря трансформации и дифференциации клеток верхней (головной) части нервного гребня, которые мигрируют вдоль нервной трубки в область первой и второй жаберных дуг зародыша. И дефицит этих клеток вызывает черепно-лицевые деформации. Критический период возникновения аномалий – с 18 по 28 день после оплодотворения. По завершении миграции клеток нервного гребня (на четвертой неделе гестации) образуются практически все рыхлые мезенхимальные ткани в области лица, которые позже (с 5 по 8 недели) дифференцируются в скелетные и соединительные ткани всех частей лица, шеи, гортани, уха (в том числе внутреннего) и будущих зубов.
, , ,
Патогенез синдрома Тричер Коллинза часто имеет семейный характер, и аномалия наследуется по аутосомно-доминантному принципу, хотя бывают случаи аутосомно-рецессивной передачи дефекта (при мутациях других генов, в частности, POLR1C и POLR1D). Самым непредсказуемым в челюстно-лицевом дизостозе является то, что мутация наследуется детьми только в 40-48% случаев. То есть у 52-60% пациентов причины синдрома Тричер Коллинза не связаны с наличием аномалии в роду, и, как полагают, патология возникает в результате спорадических генных мутаций de novo. Вероятнее всего, новые мутации представляют собой последствия тератогенного воздействия на плод во время беременности.
В числе тератогенных причин данного синдрома специалисты называют большие дозы этанола (этилового спирта), радиацию, сигаретный дым, цитомегавирус и токсоплазму, а также гербициды на основе глифосата (Раундал, Глифор, Торнадо и др.). А в список ятрогенных факторов попали препараты от угрей и себореи с 13-цис-ретиноевой кислотой (Изотретиноин, Аккутан); противосудорожное лекарство Фенитоин (Дилантин, Эпанутин); психотропные средства Диазепам, Валиум, Реланиум, Седуксен.
По большей части, клинические признаки мандибулофасциального дизостоза и степень их выраженности зависят от особенностей проявления генных мутаций. И первые признаки данной аномалии в большинстве случаев видны у ребенка сразу же после его появления на свет: лицо при синдроме Тричер Коллинза имеет характерный вид. Причем морфологические аномалии обычно двусторонние и симметричные.
Наиболее очевидные симптомы синдрома Тричер Коллинза:

Такие анатомические аномалии во всех случая имеют осложнения. Это функциональные нарушения слуха в виде проводящей (кондуктивной) тугоухости или полной глухоты; нарушения зрения из-за неправильно формирования глазных яблок; дефекты нёба вызывают трудности с кормлением и глотанием. Имеются связанные с дефектами челюстей нарушения окклюзии зубов (неправильный прикус), что, в свою очередь, вызывает проблемы с жеванием и артикуляцией. Патологии мягкого неба объясняют гнусавость голоса.
Последствия челюстно-лицевых аномалий при синдроме Тричер Коллинза проявляются в том, что при рождении ребенка его интеллектуальные способности нормальные, но из-за дефектов слуха и других нарушений отмечается вторичная задержка умственного развития.
Постнатальная диагностика синдрома Тричер Коллинза, по существу, проводится на основании клинических признаков. Челюстно-лицевой дизостоз легко определяется при полный экспрессивности синдрома, но когда присутствуют минимально выраженные симптомы патологии, с постановкой правильного диагноза могут возникнуть проблемы.
При этом особого внимания требует оценка всех связанных с аномалиями функций, особенно тех, что затрагивают дыхание (в связи с угрозой апноэ во сне). Также проводится оценка и мониторинг эффективности кормления и насыщения гемоглобина кислородом.
В дальнейшем - на 5-6 день после рождения - предстоит выяснить степень повреждений слуха с помощью аудиологического тестирования, которое должно проводиться еще в родильном доме.
Назначается обследование, в ходе которого инструментальная диагностика проводится рентгеноскопией черепно-лицевой дисморфологии; пантомографией (панорамным рентгеном костных структур лицевого черепа); полной черепной компьютерной томографией в различных проекциях; КТ или МРТ головного мозга для определения состояния внутреннего слухового прохода.
Самое раннее – пренатальное – диагностирование челюстно-лицевых аномалий при наличии синдрома Тричер Коллинза в семейном анамнезе возможно путем биопсии ворсин хориона на 10-11 неделе беременности (процедура угрожает выкидышем и занесением инфекции в матку).
Также берутся анализы крови членов семьи; на 16-17 неделе беременности берется анализ околоплодных вод (трансабдоминальный амниоцентез); на 18-20 неделях беременности проводится фетоскопия и берется кровь из плодовых сосудов плаценты.
Но чаще всего в дородовой диагностике данного синдрома у плода используется УЗИ (на 20-24 неделях беременности).
Этими же методами специалисты пользуются, когда нужна дифференциальная диагностика, чтобы распознать неярко выраженный синдром Тричер Коллинза и отличить его от других врожденных аномалий черепно-лицевых костей, в частности: синдромов Апера, Крузона, Нагера, Петерс-Хевельса, Хеллермана-Штефа, а также с гемифациальной микросомии (синдрома Гольденхара), гипертелоризма, преждевременного заращения швов черепа (краниостеноза) или нарушения сращения лицевых костей (краниосиностоза).
Синдром Франческетти является аутосомно-доминантным расстройством черепно-лицевого развития с переменной экспрессией. Широко известен как синдром Тришера Коллинза (TCS).
Назван в честь английского офтальмолога Э. Т. Коллинза, который в 1900 году описал основные компоненты этого состояния. Расстройство влияет на оба пола одинаково.
В Англии и на американском континенте эта аномалия описывается как «синдром Тришера Коллинза», на европейском континенте – «Мандибуло-лицевой дизостос» или «синдром Франческетти»
Другие названия:

Синдром Франческетти – серьезное врожденное расстройство черепно-лицевого развития, характеризующееся многочисленными аномалиями, которые ограничены головой и лицом. Гипоплазия костей лица, особенно челюстной и скуловой системы, является общей чертой.
Эта болезнь почти полностью отсекает ребенка от общества, делая его жизнь еще более тяжелой и болезненной.
Синдром Франческетти – это генетическое заболевание, вызванное мутацией генов, помогающих правильному развитию костей и лицевых тканей. Этот белок важен для развития лица как в утробе матери, так и после рождения. Мутация приводит к уменьшению его количества, изменяет клетки формирующие черты лица.
Нарушения в развитии костной ткани, вызывают сильную асимметрию, из-за чего слуховой и зрительный аппарат приобретают постоянные структурные патологии.
Редкое генетическое заболевание, затрагивает приблизительно 1 из 50 000 человек.
Развитие заболевания связано с нарушениями внутриутробного развития на 6-7 неделе.
Расстройство возникает из-за мутаций гена TCOF1 или POLR1C, POLR1D. Это аутосомно-доминантное состояние, что означает, что одна копия измененного гена в каждой клетке достаточна, чтобы вызвать расстройство.
Около 60 процентов случаев являются результатом новых мутаций и встречаются у людей, не имеющих истории семейного расстройства. Чаще, человек наследует измененный ген от пострадавшего родителя.
Когда синдром Франческетти вызывается мутациями гена POLR1C, состояние имеет аутосомно-рецессивный характер наследования. Такое наследование означает, что обе копии гена в каждой клетке имеют мутации.
Родители индивидуума с аутосомно-рецессивным состоянием имеют одну копию мутированного гена, но обычно не проявляют признаков и симптомов состояния.

Мутации гена TCOF1 являются наиболее распространенной причиной расстройства, составляя от 81 до 93 процентов всех случаев.
POLR1C и POLR1D вызывают дополнительные 2 процента случаев.
Белки, полученные из генов TCOF1, POLR1C и POLR1D, играют важную роль в раннем развитии костей и других тканей лица. Они участвуют в получении молекулы, называемой рибосомальной РНК (рРНК). Рибосомальная РНК помогает собрать белковые строительные блоки (аминокислоты) в новые белки, которые необходимы для нормального функционирования и выживания клеток.
Узнать больше Продолжительность жизни при синдроме Рубинштейна Тейби: симптомы и причины заболевания
Мутации TCOF1, POLR1C, POLR1D уменьшают продукцию рРНК. Исследователи предполагают, что уменьшение количества рРНК может спровоцировать саморазрушение (апоптоз) определенных клеток, участвующих в развитии костей и тканей лица.
Аномальная гибель клеток приводит к специфическим проблемам развития лица, обнаруженным при синдроме Франческетти.

Признаки этого расстройства сильно различаются: от почти незаметного до тяжелого. Симптомы синдрома Франческетти часто обнаруживаются на УЗИ во время беременности. Чаще проявляется деформированное и недоразвитое лицо, подбородок.
Нижние веки поражены, скулы, челюсть, уши могут отсутствовать или иметь дефекты. У детей возникают проблемы с дыханием и слухом.

Симптомы таковы:
Потеря слуха вызвана дефектами трех небольших костей в среднем ухе, которые передают звук, или из-за недостаточного развития слухового прохода.

Некоторые рождаются с отверстием в крыше рта, называемым волчьей пастью. В тяжелых случаях недоразвитие костей лица ограничивает дыхательные пути пострадавшего ребенка, вызывая потенциально опасные для жизни проблемы с дыханием.
Люди с расстройством обычно имеют нормальный интеллект.

В настоящее время нет препаратов для лечения синдрома Франческетти. Лечение направлено на конкретные потребности каждого ребенка или взрослого. Необходим многодисциплинарный медицинский подход. Главная проблема – нарушение проходимости дыхательных путей, глотание, зрение, слух.
Новорожденным требуется немедленная трахеостомия, если воздушные пути деформированы. Процедура предполагает размещение трубки непосредственно в области шеи, горла.
Потеря слуха рассматривается как усиление костной проводимости путем размещения имплантов, рекомендуется речевая терапия, образовательные программы.
Узнать больше Признаки и лечение синдрома Поланда у девочек и мальчиков
Во многих случаях необходима черепно-лицевая реконструкция. Хирургия выполняется для восстановления расщепления неба, восстановления челюсти или других костей черепа. Конкретные хирургические процедуры и возраст при проведении операции зависят от тяжести аномалий, общего состояния здоровья, личных предпочтений.
 На фотографии девушка до и после пластической хирургии. Поэтому не отчаивайтесь и не опускайте руки. Всегда возможно найти способ улучшить свой внешний вид.
На фотографии девушка до и после пластической хирургии. Поэтому не отчаивайтесь и не опускайте руки. Всегда возможно найти способ улучшить свой внешний вид. Костные трансплантаты устанавливаются, чтобы помочь создать скулы. Некоторые из операций откладываются до достижения ребенком 5 лет и старше.
В некоторых случаях невозможно полностью исправить деформации лица, поэтому врачи слегка уменьшают проявление симптомов улучшая качество жизни пациента.
Попытки восстановить слух хирургическим путем (для коррекции структуры слуховых косточек) не показали положительного результата, поэтому рекомендуется использовать слуховые аппараты, выбранные для индивидуальных патологий внутреннего и среднего уха.
Есть несколько возможных методов лечения, которые исследуются. Ученые ищут способы ингибировать протеин p53, который помогает организму убивать нежелательные клетки. У людей с синдромом Франческетти, p53 аномально активируется, что приводит к потере определенных клеток, вызывает симптоматику.
Есть предположение, что ингибирование продуцирования р53 (блокирование его активации) может помочь лечить пораженных людей. Необходимы дополнительные исследования, чтобы определить, эффективен ли этот тип лечения и безопасен ли он.
Исследователи также изучают использование стволовых клеток, обнаруженных в жировой ткани, которые используются при операциях у людей с черепно-лицевыми патологиями.
Ранние исследования показали, что хирургические результаты улучшаются с использованием этих стволовых клеток, помогают стимулировать восстановление роста пораженных участков. Однако подобная терапия все еще экспериментальная и противоречивая.