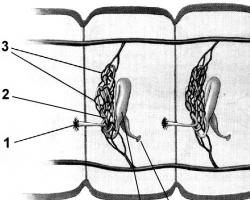
Межуточный обмен аминокислот складывается из реакций дезаминирования, трансаминирования и декарбоксилирования.
Рис. 21. Метаболизм аминокислот.
Дезаминирование. Это этап межуточного обмена аминокислот, при котором происходит образование кетокислот и аммиака. Дезаминирование осуществляется ферментом аминооксидазой, коферментом которой является ФАД или НАД.
L -глутамат → NH 3 + α -кетоглутарат
Дезаминирование является универсальным процессом в образовании аминокислот, когда неиспользованные для синтеза белка аминокислоты теряют аминогруппы и превращаются в безазотистые продукты. Из аминогруппы образуется аммиак, а из безазотистой части – кетокислоты.
Благодаря образованию α -кетоглутарата дезаминирование обеспечивает работу цикла Кребса, а благодаря образованию ионов аммония в почечных канальцах – участвует в регуляции кислотно-основного состояния (аммониогенез).
Причины и последствия недостаточности дезаминирования.
Ослаблен этот процесс при поражении печени, при гипоксии, при авитаминозах С, РР и В 2 .
Нарушение дезаминирования приводит к ослаблению мочевинообразования увеличению аминокислот в крови (аминоацидемии ), что сопровождается аминоацидурией.
Также последствиями снижения дезаминирования являются: уменьшение синтеза белка вследствие недостаточности смежных реакций трансаминирования, подавление активности цикла Кребса, энергообразования, ацидоз, гипераммониемия.
Причины и последствия избыточности дезаминирования.
Причинами увеличения дезаминирования могут быть: голодание, когда энергетические потребности организма удовлетворяются за счет белка.
Последствиями усиления дезаминирования являютсяувеличение образования α-кетоглутарата, ведущее к повышению энергообразования и образования кетокислот, уменьшение синтеза белка, повышение синтеза аммиака и увеличение мочевинообразования.
Трансаминирование (переаминирование) – это обратимый перенос аминогруппы с аминокислоты на кетокислоту без промежуточного образования аммиака с образованием новой кетокислоты (КК) и новой заменимой аминокислоты. Аминокислоты являются донаторами аминогруппы, а кетокислоты – акцепторами.
Трансаминирование протекает в присутствии кофермента, роль которого выполняет пиридоксальфосфат (витамин В 6).
Трансаминирование поставляет кетокислоты (щавелевоуксусную кислоту) в цикл Кребса, тем самым поддерживает энергетический обмен, пировиноградную кислоту – для обеспечения глюконеогенеза, синтеза заменимых аминокислот.
При переносе аминогруппы на α-кетоглутарат образуется коллекторное вещество L-глутамат:
А-та + α -кетоглутарат ↔ КК (ПК, ЩУК) + L -глутамат
L-глутамат используется в синтезе мочевины.
Причины уменьшения трансаминирования:
гиповитаминоз В 6 вследствие недостаточного содержания витамина в пище, при высокой потребности во время беременности, при нарушении его усвоения и фосфорилирования во время лечения фтивазидом, при подавлении кишечной микрофлоры, частично синтезирующей витамин, под воздействием длительного применения сульфаниламидных препаратов.
ограничение синтеза белка (при голодании и тяжелых заболеваниях печени, при недостаточности коры надпочечников и щитовидной железы).
Последствия уменьшения трансаминирования:
уменьшение синтеза заменимых аминокислот (аланина из пировиноградной кислоты, аспарагина из щавелевоуксусной кислоты);
гипогликемия вследствие уменьшения глюконеогенеза;
аминоацидемия вследствие уменьшения синтеза мочевины;
ацидоз в мышцах вследствие увеличения пировиноградной кислоты (ПК) в мышцах (из-за нарушения ее переноса в печень)
ПК + L -глутамат → α -Аланин + α -кетоглутарат
образование токсических веществ вследствие активации декарбоксилирования.
В процессе трансаминирования из триптофана образуется никотиновая кислота. Отсутствие фосфопиридоксаля приводит к нарушению синтеза никотиновой кислоты, в результате чего развивается пеллагра.
При ряде причин (избыток кетокислот (ПК, α-кетоглутарата, увеличении глюкокортикоидов) отмечается повышение трансаминирования.
Последствия повышенного трансаминирования :
уменьшение содержания незаменимых аминокислот
снижение синтеза белка,
повышение синтеза мочевины и гиперазотемия.
Если в отдельных органах возник некроз (панкреатит, гепатит, инфаркт миокарда или легких), то вследствие разрушения клеток тканевые трансаминазы поступают в кровь и повышение активности в крови является одним из диагностических тестов. Повышение уровня аспартатаминотрансферазы (АСТ) характерно для болезней сердца и аланинаминотрансферазы (АЛТ) – характерно для болезни печени.
Декарбоксилирование. Это процесс отщепления карбоксильных групп от аминокислот в виде CO 2 .
Аминокислота → Амины (биогенные) + С O 2
Первичные амины образуются при декарбоксилировании аминокислот. В эту реакцию вступают вcе аминокислоты. Процесс декарбоксилирования осуществляется специфическими декарбоксилазами, коферментом которых является фосфопиридоксаль (витамин В 6).
Декарбоксилированию с образованием биогенных аминов и углекислоты подвергаются только некоторые аминокислоты.
гистидин → гистамин
Содержание гистамина повышается при аллергических заболеваниях (бронхиальная астма, отек Квинке и др.), при ожогах, распаде опухолей, при шоках (анафилактическом, травматическом и гемотрасфузионном), при укусах ядовитых насекомых, при нервном возбуждении, кислородном голодании. Избыток гистамина повышает проницаемость сосудов, вызывает их дилатацию, нарушает микроциркуляцию, вызывает спазм гладкой мускулатуры.
триптофан → триптамин→серотонин
Серотонин образуется в митохондриях хромаффинных клеток кишечника. Разрушается в основном в легких с помощью фермента аминооксидазы. Серотонин повышает тонус гладкой мускулатуры, тонус и резистентность сосудов, является медиатором нервных импульсов в ЦНС, уменьшает агрессивность. Увеличивается содержание серотонина в крови при карциноиде кишечника, при обострении хронического панкреатита, иммобилизационном стрессе у крыс.
глутаминовая к-та → гамма-аминомасляная (ГАМК)
Гамма-аминомасляная кислота (ГАМК) тормозит синаптическую передачу поверхностных слоев коры головного мозга.
тирозин → тирамин (фальш-медиатор)
ДОФА → дофамин
цистин → таурин
Причинами повышения содержания биогенных аминов могут быть не только увеличение декарбоксилирования соответствующих аминокислот, но также угнетение окислительного распада аминов и нарушение их связи с белками. Так, например, при гипоксических состояниях, ишемии, деструкции тканей (травмы, облучение и т.д.) ослабляются окислительные процессы, что уменьшает превращение аминокислот по пути их обычного распада и усиливает декарбоксилирование.
Появление большого количества биогенных аминов в тканях (особенно гистамина и серотонина) может вызвать значительные нарушения местного кровообращения, повышение проницаемости сосудов и повреждение нервной системы.
Снижение активности декарбоксилирования отмечается при гипоксии, дефиците витамина В 6.
Гипоксия и ацидоз снижают выработку ГАМК, при дефиците которой возникают судороги, недостаточное образование нейромедиатора серотонина вызывает нарушение эмоций.
Введение…………………………………………………………………………..3
1.Наследственные болезни обмена аминокислот………………………………4
2. Наследственные нарушения обмена аминокислот…………………………..5
3. Фенилкетонурия………………………………………………………………..6
4. Клинические симптомы у больных фенилкетонурией………………………8
5. Гомоцистинурия………………………………………………………………11
6. Гистидинемия…………………………………………………………………15
7. Наследственные нарушения обмена триптофана…………………………...17
8. Галактоземия…………………………………………………………………..19
9. Недостаточность лактазы…………………………………………………….22
10. Врожденные нарушения обмена гликогена………………………………..24
Заключение……………………………………………………………………….33
Список литературы………………………………………………………………34
Введение
В последние десятилетия научный прогресс в области клинической и молекулярной генетики, биохимии позволил выявить обширную группу “новых” болезней детского возраста, связанных с нарушением обмена веществ. Патологии обмена веществ у взрослых и детей могут быть обусловлены наследственными дефектами обмена нуклеиновых кислот, врожденной недостаточностью ферментов, отвечающих за синтез и распад аминокислот, нарушениями обмена органических кислот, дефицитом жирных кислот и др. Клинический диагноз врожденных нарушений обмена веществ может представлять определенные трудности. Одна из трудностей ранней диагностики заключается в том, что в период новорожденности у этих детей нет специфических расстройств, а поздние проявления фенотипически схожи с заболеваниями ненаследственного генеза. Вторая особенность состоит в том, что для наследственных заболеваний обмена веществ характерен клинический полиморфизм, обусловленный генетической гетерогенностью. Это объясняется наличием множественных изоаллельных мутаций и возможностью возникновения мутаций в разных генах.
Клинические проявления наследственных болезней обмена веществ во многом определяются поражением нервной системы (особенно при нарушениях обмена аминокислот, липидов и кислых гликозамино-гликанов), что в свою очередь, усиливает имеющиеся нарушения и усугубляет тяжесть клинических проявлений заболевания. Для диагностики наследственных болезней важен анализ неврологических симптомов, особенно на ранних стадиях развития, и разграничение их от фенокопий - заболеваний ненаследственной природы со сходной клинической картиной.
Наследственные болезни обмена аминокислот
Роль аминокислот для организма человека чрезвычайно велика. Аминокислоты являются основными структурными элементами белков, необходимы для синтеза иммуноглобулинов, гормонов, служат источником энергии. Каждый фермент или белок имеет специфические свойства и функции, которые определяют и регулируют сложные обменные процессы и развитие организма.
Часть аминокислот не может синтезироваться в организме человека. Это незаменимые аминокислоты: триптофан, фенилаланин, метионин, лизин, лейцин, изолейцин, валин и треонин. В детском возрасте к их числу относится гистидин, т.к. организм ребенка не может синтезировать эту аминокислоту в необходимых для нормального роста количествах. Клетки растущих тканей содержат аминокислоты в высоких концентрациях, что является свидетельством высокой интенсивности процессов транспорта аминокислот через клеточные мембраны.
Для обеспечения нормального роста и развития важно не только количество поступающих аминокислот, но и их соотношение. При избытке или недостатке аминокислот развиваются явления аминокислотного дисбаланса. Например, избыток лейцина в пище тормозит рост организма, метионина- вызывает токсическое поражение нервной системы, цистина- способствует развитию жировой инфильтрации печени.
Таким образом, нарушения метаболизма аминокислот приводят к нарушению нормального функционирования организма человека.
Наследственные нарушения обмена аминокислот
1. Наследственные нарушения обмена аминокислот, сопровождающиеся увеличением их концентрации в крови и моче: фенилкетонурия, гистидинемия, триптофанурия, болезнь "кленового сиропа", орнитинемия, цитруллинемия и др. Наследование, в основном, по аутосомно-рецессивному типу. В основе развития заболеваний лежит нарушение синтеза или структуры тех или иных ферментов.
2. Наследственные нарушения обмена аминокислот, сопровождающиеся увеличением их выделения с мочой без изменения уровня в крови: гомоцистинурия, гипофосфатазия и др. При данных энзимопатиях нарушено обратное всасывание в почках, что приводит к увеличению их содержания в моче.
3. Наследственные нарушения систем транспорта аминокислот: цистинурия, триптофанурия, болезнь Гартнепа и др. К этой группе относятся энзимопатии, развитие которых обусловлено снижением реабсорбции аминокислот в почках и кишечнике.
4. Вторичные гипераминоцидурии: синдром Фанкони, фруктоземия, галактоземия, болезнь Вильсона-Коновалова и др. При данных состояниях возникает вторичная генерализованная гипераминоацидурия в результате вторичных тубулярных нарушений.
Фенилкетонурия (ФКУ)
Впервые описана в 1934 г. Folling под названием "фенилпировиноградная имбецильность". Тип наследования - аутосомнорецессивный. Частота заболевания составляет 1:10000- 1:20000 новорожденных. Пренатальный диагноз возможен при использовании генетических зондов и биопсии ворсин хориона.
К развитию классической клинической картины при ФКУ приводит недостаточность фенилаланингидроксилазы и недостаточность редуктазы дигидроптерина- 2-го фермента, обеспечивающего гидроксилирование фенилаланина. Их недостаток приводит к накоплению фенилаланина (ФА) в жидких средах организма (схема 1). Как известно, ФА относится к незаменимым аминокислотам. Поступающий с продуктами питания и не используемый для синтеза белка, он распадается по тирозиновому пути. При ФКУ наблюдается ограничение превращения ФА в тирозин и, соответственно, ускорение его превращения в фенилпировиноградную кислоту и другие кетоновые кислоты.
Схема 1. Варианты нарушений метаболизма фенилаланина.
Существование различных клинико-биохимических вариантов ФКУ объясняется тем, что фенилаланингидроксилаза является частью мультиферментной системы.
Различают следующие формы ФКУ:
1.Классическая
2.Скрытая.
3.Атипичная.
Развитие атипичных и скрытых форм ФКУ связывают с недостаточностью фенилаланинтрансаминазы, тирозинтрансаминазы и оксидазы парагидроксифенилпировиноградной кислоты. Атипичная ФКУ обычно не сопровождается поражением нервной системы в результате позднего развития ферментативного дефекта.
У женщин с фенилкетонурией возможно рождение детей с микроцефалией, задержкой умственного развития, нарушениями развития мочевыделительной системы, поэтому необходимо назначение диетотерапии во время беременности.
Нарушение трансаминирования и окислительного дезаминирования. Процессы трансаминирования и дезаминирования имеют универсальное значение для всех живых организмов: трансаминирование способствует синтезу аминокислот, дезаминирование - их разрушению.
Суть реакции трансаминирования состоит в обратном переносе аминогруппы с аминокислоты в α-кетокислоту без промежугочного образования свободного иона аммония. Реакция катализируется специфическими ферментами аминотрансферазами (трансаминазами), кофакторами которых являются фосфорилированные формы пиридоксина (пиридоксальфосфат и пиридоксаминфосфат).
Нарушения реакций трансаминирования могут возникать по нескольким причинам, прежде всего - в результате дефицита пиридоксина (беременность, угнетение сульфаниламидными препаратами кишечной микрофлоры, торможение синтеза пиридоксальфосфата при лечении фтивазидом). Снижение активности аминотрансфераз происходит также в случае угнетения синтеза белков (голодание, тяжелая патология печени). Если в некоторых органах возникает некроз (инфаркт миокарда или легких, панкреатит, гепатит и др.), то вследствие разрушения клеток тканевые аминотрансферазы поступают в кровь, и повышение их активности в крови при такой патологии является одним из диагностических критериев. В изменении скорости трансаминирования важную роль играют нарушение соотношения субстратов реакции, а также влияние гормонов, особенно глюкокортикоидов и гормонов щитовидной железы, стимулирующих этот процесс.
Угнетение процесса окислительного дезаминирования, в результате которого распадаются неиспользованные аминокислоты, обусловливает повышенную концентрацию их в крови - гипераминоацидемию . Последствиями этого являются усиленная экскреция аминокислот почками (аминоацидурия ) и изменение соотношения отдельных аминокислот в крови, что создает неблагоприятные условия для синтеза белковых молекул. Дезаминирование нарушается при дефиците компонентов, которые прямо или косвенно принимают участие в этой реакции (пиридоксин, рибофлавин, никотиновая кислота), а также при гипоксии, голодании (белковая недостаточность).
Нарушение декарбоксилирования. Этот процесс является важным, хотя и не универсальным направлением белкового обмена, и происходит с образованием углекислого газа и биогенных аминов. Декарбоксилированию подвергаются лишь некоторые аминокислоты: гистидин преобразуется в гистамин, тирозин - в тирамин, γ-глугаминовая кислота - в γ-аминомасляную кислоту (ГАМК), 5-гидрокситриптофан - в серотонин, производные тирозина (3,4-диоксифенилаланин) и цистина (L-цистеиновая кислота - соответственно в 3,4-диоксифенилэтиламин (дофамин) и таурин.
Биогенные амины, как известно, имеют специфическую биологическую активность, и увеличение их количества может вызвать определенные патологические изменения в организме. Большое количество биогенных аминов может быть результатом не только усиленного декарбоксилирования соответствующих аминокислот, но и угнетения окисления аминов и нарушения связывания их белками. Например, при гипоксии, ишемии и деструкции тканей (травма, облучение и т. п.) замедляются окислительные процессы, тем самым способствуя усилению декарбоксилирования. Избыток биогенных аминов (особенно гистамина и серотонина) в тканях может обусловить значительное нарушение местного кровообращения, повышение проницаемости сосудистой стенки и повреждение нервного аппарата.
Наследственные нарушения обмена некоторых аминокислот
Метаболизм аминокислот детерминируется определенным количеством и активностью соответствующих ферментов. Наследственные нарушения синтеза ферментов приводят к тому, что необходимая аминокислота не включается в метаболизм, а накапливается в биологических средах организма: крови, моче, кале, поту, спинномозговой жидкости. Клиническая картина в таких случаях обусловлена, во-первых, наличием достаточно большого количества вещества, которое должно было метаболизоваться с помощью заблокированного фермента; во-вторых - дефицитом вещества, которое должно было образоваться.
Генетически обусловленных нарушений обмена аминокислот известно довольно много, все они наследуются по аутосомно-рецессивному типу. Некоторые из них приведены в табл. 2.
Нарушение обмена фенилаланина.
В норме фенилаланин преобразуется в тирозин. Если в печени нарушается синтез необходимого для этого фермента фенила-ланингидроксилазы (схема 4), то окисление фенилаланина происходит посредством образования фенилпировиноградной и фенилмолочной кислот - развивается фенилкетонурия. Однако этот путь имеет малую “пропускную” способность, поэтому большое количество фенилаланина накапливается в крови, тканях и спинномозговой жидкости, что в первые же месяцы жизни новорожденного проявляется тяжелым поражением ЦНС и неизлечимым слабоумием. Вследствие недостаточного синтеза тирозина угнетается образование меланина, который обусловливает осветление кожи и волос. Кроме того, в результате повышенного образования фенилпировиноградной кислоты тормозится активность фермента дофамингидроксилазы, необходимого для синтеза катехоламинов (адреналина, норадреналина). Тяжесть наследственной патологии определяется комплексом всех этих нарушений. Больные умирают в детстве, если не проводится специальное лечение, заключающееся в постоянном, но осторожном (контроль аминокислотного состава крови) ограничении поступления фенилаланина с пищей. Раннюю диагностику заболевания нужно проводить сразу после рождения ребенка. Для этого применяют различные биохимические тест-системы.
Нарушение обмена тирозина.
Обмен тирозина происходит несколькими путями. В случае недостаточного преобразования тирозина в гомогентизиновую кислоту (см. схему 4), что может быть обусловлено дефектом различных ферментов, тирозин накапливается в крови и выводится с мочой. Это нарушение называется тирозинозом и сопровождается печеночной и почечной недостаточностью и ранней смертью ребенка или лишь задержкой психомоторного развития. Если нарушение обмена тирозина происходит в момент окисления гомогентизиновой кислоты (см. схему 4), развивается алкаптонурия. Фермент, окисляющий гомогентизиновую кислоту (гомогентизиноксидаза), образуется в печени. В норме он настолько быстро разрывает ее гидрохиноновое кольцо, что кислота “не успевает” попасть в кровь, а если и попала, то быстро выделяется почками. В случае наследственного дефекта этого фермента гомогентизиновая кислота в большом количестве накапливается в крови и моче. Моча больных алкаптонурией на воздухе или после добавления щелочи становится черной. Это объясняется окислением гомогентизиновой кислоты кислородом воздуха и образованием в ней алкаптона (от лат. alcapton - захватывающий щелочь). Гомогентизиновая кислота с током крови поступает в ткани - хрящевую, сухожилия, связки, внутренний слой стенки аорты, вследствие чего образуются темные пятна в области ушей, носа, щек, на склерах. Алкаптон делает хрящи и сухожилия хрупкими, что иногда приводит к тяжелым изменениям в суставах.
Также тирозин - это исходный продукт для образования пигмента меланина, содержащегося в коже и волосах. Если преобразование тирозина в меланин замедленно вследствие наследственного дефицита тирозиназы (см. схему 4), возникает альбинизм , который сопровождается повышением чувствительности кожи к солнечному свету и нарушением зрения.
И наконец, тирозин является предшественником тироксина. В случае недостаточного синтеза фермента, который катализирует взаимодействие тирозина со свободным йодом, нарушается образование гормонов щитовидной железы.
Нарушение обмена триптофана. Основной путь метаболизма триптофана, как и никотиновой кислоты, обеспечивает синтез никотинамидадениндинуклеотида (НАД) и НАДФ, которые играют важную роль в жизнедеятельности организма, будучи коферментами многих реакций обмена, а значительный дефицит этих веществ служит причиной развития пеллагры . Нарушение обмена триптофана также может сопровождаться изменением количества образующегося из него серотонина.
Генные болезни человека
Генные болезни – это разнообразная по клинической картине группа заболеваний, обусловленная мутациями единичных генов.
Число известных в настоящее время моногенных наследственных заболеваний составляет около 4500. Встречаются эти заболевания с частотой 1: 500 - 1: 100000 и реже. Моногенная патология определяется примерно у 3% новорожденных и является причиной 10% младенческой смертности.
Наследуются моногенные заболевания в соответствии с законами Менделя.
Начало патогенеза любой генной болезни связано с первичным эффектом мутантного аллеля. Он может проявляться в следующих вариантах: отсутствие синтеза белка; синтез аномального белка; количественно избыточный синтез белка; количественно недостаточный синтез белка.
Патологический процесс, возникающий в результате мутации единичного гена, проявляется одновременно на молекулярном, клеточном и органном уровнях у одного индивида.
Существует несколько подходов к классификации моногенных болезней: генетический, патогенетический, клинический и др.
Классификация, основанная на генетическом принципе: согласно ей моногенные болезни можно подразделять по типам наследования – аутосомно-доминантные, аутосомно-рецессивные, Х-сцепленные доминантные, Х-сцепленные рецессивные, У-сцепленные (голандрические). Эта классификация наиболее удобна, т.к. позволяет сориентироваться относительно ситуации в семье и прогноза потомства.
Вторая классификация основана на клиническом принципе, т.е. на отнесении болезни к той или иной группе в зависимости от системы органов, наиболее вовлеченной в патологический процесс, - моногенные заболевания нервной, дыхательной, сердечно-сосудистой систем, органов зрения, кожи, психические, эндокринные и т.д.
Третья классификация основывается на патогенетическом принципе. Согласно ей все моногенные болезни можно разделить на три группы:
наследственные болезни обмена веществ;
моногенные синдромы множественных врожденных пороков развития;
комбинированные формы.
Рассмотрим наиболее распространенные моногенные заболевания.
Нарушение обмена аминокислот.
Наследственные заболевания, обусловленные нарушением обмена аминокислот, составляют значительную часть генетической патологии детей раннего возраста. Большинство из них начинаются после достаточно короткого периода благополучного развития ребёнка, но в дальнейшем приводят к тяжелому поражению интеллекта и физических показателей. Встречается и острое течение этих заболеваний, когда состояние новорожденного резко ухудшается на 2-5-е сутки жизни. В такой ситуации высока вероятность летального исхода ещё до момента уточнения диагноза.
Абсолютное большинство этих болезней наследуется аутосомно-рецессивно. Вероятность повторного рождения больного ребёнка в семьях, где уже регистрировалась эта патология, составляет 25%.
Фенилкетонурия (ФКУ) – самое распространенное заболевание, вызванное нарушением аминокислотного обмена. Впервые было описано в 1934 году. Это заболевание наследуется аутосомно-рецессивно.
В Западной Европе один больной ФКУ обнаруживается среди 10000-17000 новорожденных, в Беларуссии и России частота ФКУ колеблется в пределах 1 случай на 6000-10000 новорожденных. Очень редко ФКУ встречается среди негров, евреев-ашкеназов, в Японии.
Основной причиной ФКУ является дефект фермента фенилаланин-4-гидроксилазы, который способствует превращению аминокислоты фенилаланина в тирозин. Фенилаланин относится к жизненно необходимым аминокислотам, которые не синтезируются в организме, а поступают с продуктами питания, содержащими белок. Фенилаланин входит в состав многих белков человека, имеет большое значение для созревания нервной системы.
Ген, определяющий структуру фенилаланин-4-гидроксилазы, локализован на длинном плече 12-й хромосомы, содержит 70000 пар нуклеиновых оснований. Чаще всего мутация этого гена вызвана заменой одного нуклеотида (90% всех случаев заболевания).
Дефект фермента при ФКУ приводит к нарушению реакции превращения фенилаланина в тирозин. В результате в организме больного накапливается избыточное количество фенилаланина и его производных: фенилпировиноградной, фенилмолочной, фенилуксусной и др. В то же время при ФКУ в организме больного формируется недостаток продуктов реакции: тирозина, являющегося важной частью обмена нейромедиаторов (катехоламинов и серотонина), и меланина, определяющего окрашивание кожи и волос у человека.
Избыток фенилаланина и его производных оказывает непосредственное повреждающее действие на нервную систему, функцию печени, обмена белков и других веществ в организме.
Беременность и роды при ФКУ у плода обычно не имеют каких-либо специфических особенностей. Новорожденных ребёнок выглядит здоровым, так как в период в период внутриутробного развития обмен веществ матери обеспечивает нормальный уровень фенилаланина в организме плода. После рождения ребенок начинает получать белок с молоком матери. Дефект фенилаланингидроксилазы препятствует обмену содержащегося в белке грудного молока фенилаланина, который начинает постепенно накапливаться в организме больного.
Первые клинические проявления ФКУ можно заметить у 2-4-месячного ребенка. Кожа и волосы начинают терять пигментацию. Глаза становятся голубыми. Часто появляются экземоподобные изменения кожных покровов: покраснения, мокнутие и шелушение щечек и складок кожи, коричневатые корочки в области волосистой части черепа. Возникает, а затем усиливается специфический запах, описываемый как «мышиный».
Ребёнок становится вялым, теряет интерес к окружающему. С 4 месяцев становится заметной задержка моторного и психического развития. Ребёнок значительно позже начинает сидеть, ходить, не всегда способен научиться разговаривать. Степень выраженности поражения нервной системы варьирует, но при отсутствии лечения обычно регистрируется глубокая умственная отсталость. Примерно у четверти больных детей во втором полугодии жизни возникают судороги. Особенно характерны кратковременные приступы, сопровождающиеся наклонами головы («кивки»). Дети с ФКУ старше 1 года обычно расторможены, эмоционально неустойчивы.
Диагностика ФКУ основывается не только на клиническом осмотре и генеалогических данных, но и на результатах лабораторных исследований (определение фенилпировиноградной кислоты в моче). Для уточнения диагноза необходимо определение уровня фенилаланина в крови ребенка (в норме содержание фенилаланина в крови не более 4 мг%, у больного ФКУ превышает 10, а иногда и 30 мг%).
Поскольку главной причиной поражения нервной системы при классической форме ФКУ является избыток фенилаланина, то ограничение его поступления с пищей в организм больного даёт возможность предупредить патологические изменения. С этой целью применяется специальная диета, обеспечивающая только минимальную возрастную потребность в фенилаланине для ребенка. Эта аминокислота входит в структуру большинства белков, поэтому из рациона больного исключаются высокобелковые продукты: мясо, рыба, творог, яичный белок, хлебобулочные изделия и др.
Раннее введение диеты (на 1-ом месяце жизни) и её регулярное соблюдение обеспечивает практически нормальное развитие ребенка.
Строгая диетотерапия рекомендуется до 10-12 лет. После этого объем обычных продуктов питания для больных ФКУ постепенно увеличивается, и пациенты переводятся на вегетарианское питание. В случае повышенной физической или умственной нагрузки рекомендуют использовать в пищу заменители белка.
В зрелом возрасте строгая диета необходима женщинам, больным ФКУ, которые планируют деторождение. Если уровень ФА крови беременной превышает нормальный, то её ребёнок будет иметь микроцефалию, врожденный порок сердца и другие аномалии.
Нарушение обмена соединительной ткани.
Абсолютное большинство этих болезней наследуется аутосомно-доминантно. При данном типе наследования больные встречаются в каждом поколении; у больных родителей рождается больной ребёнок; вероятность наследования составляет 100% - если хотя бы один родитель гомозиготен, 75% - если оба родителя гетерозиготны, и 50% - если один родитель гетерозиготен.
Синдром Марфана. Это одна из наследственных форм врожденной генерализованной патологии соединительной ткани, впервые описана в 1886 году В. Марфаном. Частота в популяции – 1: 10000-15000.
Этиологическим фактором синдрома Марфана (СМ) является мутация в гене фибриллина, локализованном в длинном плече 15-й хромосомы.
Больные синдромом Марфана имеют характерный внешний вид: они отличаются высоким ростом, астеническим телосложением, количество подкожно-жировой клетчатки у них снижено, конечности удлинены преимущественно за счет дистальных отделов, размах рук превышает длину тела (норме эти показатели совпадают). Отмечаются длинные тонкие пальцы – паукообразные (арахнодактилия), часто наблюдается «симптом большого пальца», при котором длинный 1-ый палец кисти в поперечном положении достигает ульнарного края узкой ладони. При охватывании 1-ым и 5-м пальцами запястья другой руки они обязательно перекрываются (симптом запястья). У половины больных отмечается деформация грудной клетки (воронкообразная, килеобразная), искривление позвоночника (кифоз, сколиоз), гиперподвижность суставов, клинодактилия мизинцев, сандалевидная щель. Со стороны сердечно-сосудистой системы наиболее патогномоничными являются расширение восходящей части аорты с развитием аневризмы, пролапс сердечных клапанов. Со стороны органов зрения наиболее характерны подвывихи и вывихи хрусталиков, отслойка сетчатки, миопия, гетерохромия радужки. У половины больных отмечаются паховые, диафрагмальные, пупочные и бедренные грыжи. Может наблюдаться поликистоз почек, нефроптоз, понижение слуха, глухота.
Психические и умственное развитие больных не отличается от нормы.
Прогноз жизни и здоровья определяется прежде всего состоянием сердечно-сосудистой системы. Средняя продолжительность жизни при выраженной форме синдрома Марфана около 27 лет, хотя часть больных доживает до глубокой старости.
При ведении беременных с СМ необходимо помнить о возможности расслоения аневризмы аорты и последующего её разрыва. Эти осложнения возникают обычно на поздних стадиях беременности.
Синдромом Марфана страдали президент США Авраам Линкольн, скрипач Николо Паганини.
Нарушение обмена углеводов.
Эти заболевания развиваются при врожденной недостаточности ферментов или транспортных систем мембран клеток, которые необходимы для обмена какого-либо углевода.
Клинические проявления этих патологических состояний очень разнообразны. Но для многих из них характерно начало болезни после того, как в организм ребёнка попадает соответствующий углевод. Так, галактоземия развивается с первых дней жизни ребёнка после того, как он начинает питаться молоком, фруктоземия – обычно после введения соков, сахара и блюд прикорма. Нарушение обмена углеводов часто сопровождается нарушением их всасывания в кишечнике (синдром мальабсорбции). Накапливающийся в просвете кишки сахар увеличивает содержание воды в тонком кишечнике. Всё это приводит к диарее (поносам), вздутию и болям в животе, срыгиванию.
Однако при дефектах обмена углеводов определяется поражение и других органов: нервной системы, печени, глаз и т.д.
Эти заболевания встречаются относительно редко. Исключением является врожденная лактазная недостаточность.
Галактоземия – это патология впервые была описана в 1908 году. Ген этого заболевания локализован на коротком плече 9-й хромосомы.
Причиной классической формы галактоземии является дефицит фермента галактозо-1-фосфоуридилтрансферазы, который приводит к накоплению в тканях больного ребёнка галактозо-1-фосфата. Это заболевание наследуется по аутосомно-рецессивному типу и встречается с частотой 1: 15000-50000.
Галактоза – основной фермент молока, в том числе и женского. Поэтому патологические изменения возникают с первых дней жизни ребёнка, как только он начинает вскармливаться молоком.
Сначала появляется рвота, диарея, желтушность кожи, которая не исчезает и после периода новорожденности. В дальнейшем увеличивается печень и селезенка. При приёме молочной пищи у ребенка регистрируется низкий уровень глюкозы в крови. В первые месяцы жизни ребёнка формируется помутнение хрусталиков глаз (катаракта), нарушаются функции почек. Постепенно становится заметной задержка умственного и физического развития, возможно возникновение судорог, даже смерть ребёнка на фоне очень низкого уровня глюкозы в крови или цирроза печени.
Главным в лечении этого дефекта обмена является назначение специальной диеты, не содержащей продуктов с галактозой. Раннее начало подобной терапии предупреждает поражение печени и почек, тяжелые неврологические изменения у таких больных. Возможно рассасывание катаракты. Уровень глюкозы крови нормализуется. Однако даже у пациентов, которые получают специальную диету с периода новорожденности, могут регистрироваться некоторые признаки поражения нервной системы и гипофункция яичников у девочек.
В настоящее время известны и другие типы галактоземии, которые не сопровождаются тяжелым нарушением состояния здоровья. Так, при атипичных вариантах заболевания, связанных с дефицитом галактокиназы и уридиндифосфогалактозо-4-эпимеразы, клинические проявления обычно отсутствуют. При недостаточности фермента галактокиназы единственным симптомом является катаракта. Поэтому у детей с врожденной катарактой необходимо исследовать уровень галактозы в моче и крови. При этом заболевании рано начатая диетотерапия тоже способствует восстановлению прозрачности хрусталика.
Нарушение обмена гормонов.
Врожденный гипотиреоз – один из самых распространенных дефектов обмена веществ. Это заболевание обнаруживается примерно у 1 на 4000 новорожденных Европы и Северной Америки. Несколько чаще эта патология встречается у девочек.
Причиной заболевания является полная или частичная недостаточность гормонов щитовидной железы (тиреоидных), которая сопровождается снижением скорости обменных процессов в организме. Подобные изменения приводят к торможению роста и развития ребёнка.
Врожденный гипотиреоз разделяют на первичный, вторичный и третичный.
Первичный гипотиреоз составляет около 90% всех случаев заболевания. Причиной его является поражение самой щитовидной железы. В большинстве случаев обнаруживается её отсутствие (аплазия) или недоразвитие (гипоплазия). Часто щитовидная железа оказывается не в обычной месте (на корне языка, в трахее и т.д.) Эта форма заболевания обычно регистрируется как единственный случай в семье. Однако описаны аутосомно-рецессивный и аутосомно-доминантный типы наследования порока развития щитовидной железы.
Примерно 10% всех случаев первичного гипотиреоза обусловлены дефектом образования гормонов. При этой форме заболевания отмечается увеличение размеров щитовидной железы у ребёнка (врожденный зоб). Данная патология наследуется аутосомно-рецессивно.
Вторичный и третичный гипотиреоз регистрируется только в 3-4% случаев. Эти формы заболевания обусловлены нарушением функции гипофиза и гипоталамуса, наследуется аутосомно-рецессивно.
В последние годы описаны случаи врожденного гипотиреоза, вызванные нечувствительностью тканей к действию тиреоидных гормонов. Это нарушение также характеризуется аутосомно-рецессивным типом наследования.
Недостаток тиреоидных гормонов приводит к задержке дифференцировки мозга, уменьшению количества нейронов, нейромедиаторов и других веществ. Все это вызывает угнетение функции ЦНС и задержку психического развития ребенка.
Кроме того, при гипотиреозе снижается активность ферментных систем, скорость окислительных процессов, происходит накопление недоокисленных продуктов обмена. В результате замедляется рост и дифференцировка практические всех тканей организма ребёнка (скелета, мышц, сердечно-сосудистой и иммунной систем, эндокринных желез и т.д.)
Клиническая картина всех форм гипотиреоза практически однотипна. Различается только степень тяжести заболевания. Возможно как легкое, малосимптомное течение при частично сохраненной функции тиреоидных гормонов, так и очень тяжелое состояние больного.
Врожденный гипотиреоз развивается постепенно в течение первых месяцев жизни ребенка. Несколько позже заболевание проявляется у детей, находящихся на естественном вскармливании, так как грудное молоко содержит тиреоидные гормоны.
У 10-15% больных детей первые признаки гипотиреоза можно обнаружить уже на первом месяце жизни. Роды таким ребёнком обычно происходят позже 40 недель (переношенная беременность). Новорожденные с этим заболеванием имеют большую массу тела, часто выше 4 кг. При осмотре такого ребёнка можно отметить отёчность тканей лица, большой язык, лежащий на губах, отёки в виде «подушечек» на тыльной поверхности кистей и стоп. В дальнейшем наблюдается грубый голос при плаче.
Больной ребёнок плохо удерживает тепло, вяло сосёт. Часто желтушность кожи сохраняется до 1 месяца и более.
Полного развития клиническая картина обычно достигает к 3-6 месяцам. Ребенок начинает отставать в росте, плохо набирает массу тела, лениво сосет. Кожа больного становится сухой, желтовато-бледной, утолщенной, часто шелушится. Обнаруживается большой язык, низкий хриплый голос, ломкие, сухие волосы, обычно холодные кисти и стопы, запоры. Мышечный тонус снижен. В этот период формируются особенности лицевого скелета: широкая запавшая переносица, широко расставленные глаза, низкий лоб.
После 5-6 месяцев становится заметной нарастающая задержка психомоторного и физического развития больного ребенка. Ребенок значительно позже начинает сидеть, ходить, формируется умственная отсталость. Изменяются пропорции скелета: укорачивается шея, конечности и пальцы, усиливаются грудной кифоз и поясничный лордоз, кисти и стопы становятся широкими. Ребенок начинает значительно отставать в росте. Сохраняются и усугубляются деформация лица, восковая бледность и утолщение кожи, низкий грубый голос. Мышечный тонус снижен. Больные страдают запорами. При осмотре обращается внимание на увеличение камер сердца, глухость его тонов, брадикардию, вздутый живот, пупочные грыжи. Лабораторное исследование обнаруживает нарушение возрастной дифференцировки скелета, анемию, гиперхолестеринемию.
Диагноз гипотиреоза подтверждается исследованием тиреотропного гормона гипофиза (ТТГ), тиреоидных гормонов: трийодтиронина (ТЗ) и тироксина (Т4) крови. Для больных характерно снижение уровня Т3 и Т4 крови. Уровень ТТГ увеличен при первичной форме заболевания и является низким при вторичном и третичном гипотиреозе.
Главным в лечении детей с врожденным гипотиреозом является постоянная, пожизненная терапия препаратами гормонов щитовидной железы. Если ребенок начинает принимать эти лекарственные средства на первом месяце жизни, то возможно обратное развитие всех патологических изменений в нервной системе. При условии раннего начала лечения и постоянного приема необходимой дозы тиреоидных гормонов под контролем их содержания в крови в абсолютном большинстве случаев психомоторное и физическое развитие больных детей оказывается в пределах нормы.
Особенности ухода за больными с наследственной патологией.
Пациенты, имеющие наследственную патологию, нуждаются в постоянном наблюдении медицинских работников. Хронические прогрессирующее течение заболевания делает необходимым длительное пребывание в стационарах разного профиля, частые обращения в амбулаторные учреждения.
Уход за такими больными представляет собой сложную задачу. Часто приходится иметь дело не с одним человеком, а с целой семьей, так как даже физически здоровые родственники могут нуждаться в психологической поддержке, помощи, а иногда и в превентивном лечении.
Режим дня больного с наследственной патологией должен быть по возможности приближен к обычному для соответствующего возраста. Организация прогулок, игр, учёбы, общения со сверстниками способствуют социальной адаптации больных и их семей. При заболеваниях, характеризующихся нарушением умственного развития, важно обеспечить частое общение с ребёнком, разнообразие игрушек и пособий, развивающие занятия. Формированию моторных навыков помогают регулярные занятия лечебной физкультурой и массажем.
Питание больных должно быть сбалансировано по основным ингредиентам и соответствовать возрасту. В случаях необходимости кормления через зонд при нарушении жевания и глотания дети должны получать протертое мясо, овощи и фрукты в соответствии с возрастом, а не только молоко и каши. Если такой ребёнок будет вскармливаться только молоком и кашами, он будет отставать по массе и длине тела, возникнет анемия и иммунодефицитное состояние.
Особого внимания заслуживает специальная диетотерапия при некоторых заболеваниях обмена веществ (фенилкетонурии, галактоземии, гиперхолестеринемии и т.д.) Необходима постоянная помощь родителям и семьям больных в организации питания. Кроме того, подобная диетотерапия должна сопровождаться регулярным контролем показателей массы и длины тела ребёнка: на 1-м голу жизни – ежемесячно, до трех лет – 1 раз в 3 месяца до подросткового возраста – каждое полугодие.
Дети с наследственной патологией часто страдают нарушением естественных отправлений. Для предупреждения запоров в питание больных вводят продукты, богатые клетчаткой, соки. При отсутствии самостоятельного стула нужно поставить очистительную клизму. Некоторые болезни обмена веществ и пороки развития органов желудочно-кишечного тракта сопровождаются учащенным стулом. В таких случаях нужно особенно тщательно следить за сухостью кожи ребёнка. Каждый раз ребенка необходимо обмыть теплой водой, промокнуть кожу мягкой салфеткой и обработать складки кожи растительным маслом или детским кремом.
Наследственные заболевания могут сопровождаться нарушением мочеиспускания. При такой патологии проводится учёт количества выпитой жидкости. При атонии мочевого пузыря, вызванной поражением нервной системы, используется его катетеризация.
Больные с наследственной патологией нуждаются в создании оптимальных условий по температуре и влажности в помещениях, где они находятся, поскольку такие дети часто страдают нарушением терморегуляции и склонны к перегреванию и переохлаждению.
Кроме того, комнаты, в которых ребенок проводит время, должны быть освобождены от опасных предметов (колющих, режущих, очень горячих и т.д.)
Пациенты, вынужденные длительное время проводить в лежащем положении, могут иметь пролежни. С целью их предупреждения необходимы: частая смена нательного и постельного белья; разглаживание складок на ткани, соприкасающейся с кожей больного; использование специальных подкладочных резиновых кругов или тканевых матрасов; систематическая смена положения тела больного. В таких случаях кожу больного необходимо обрабатывать камфорным спиртом или одеколоном 2-3 раза в день и затем присыпать тальком.
Важнейшей частью ухода за пациентами с наследственной патологией является работа с их родственниками. Доброжелательное отношение к больному, разъяснение родителям сущности заболевания, освобождение их от чувства вины перед ребенком, создание положительной установки на лечение – все это снижает тревожность в семье и улучшает результаты реабилитационных мероприятий.
Книга >> Медицина, здоровье... другие аллергические болезни дыхательной системы. Хронические обструктивные болезни ... нарушениями обмена аминокислот , липидов, углеводов, соединительной ткани . Подходы к лечению наследственных нарушений метаболизма. Генная ... обмен в организме человека 4 2 2 ...
... человека . Классификация болезней человека (... обмена или морфогенетических процессов. Другая черта клинической картины генных болезней ... тканей -мишеней... нарушениями обмена пуринов. Болезнь ... аминокислот в белке, можно «воссоздать» последовательность гена ...
У человек составляют генные болезни . Эти болезни наследуются... группу наследственных болезней обмена составляют нарушения обмена аминокислот В настоящее... соединительной ткани в связи нарушениями синтеза коллагена, переразгибание коленного, локтевого и других
(Ю.И. Барашнев, Ю.Е. Вельтищев, 1978 г.)
1. Наследственные нарушения обмена аминокислот, сопровождающиеся увеличением их концентрации в крови и моче: фенилкетонурия, гистидинемия, триптофанурия, болезнь "кленового сиропа", орнитинемия, цитруллинемия и др. Наследование, в основном, по аутосомно-рецессивному типу. В основе развития заболеваний лежит нарушение синтеза или структуры тех или иных ферментов.
2. Наследственные нарушения обмена аминокислот, сопровождающиеся увеличением их выделения с мочой без изменения уровня в крови: гомоцистинурия, гипофосфатазия, аргиносукцинатацидурия и др. При данных энзимопатиях нарушено обратное всасывание в почках, что приводит к увеличению их содержания в моче.
3. Наследственные нарушения систем транспорта аминокислот: цистинурия, триптофанурия, болезнь Гартнепа и др. К этой группе относятся энзимопатии, развитие которых обусловлено снижением реабсорбции аминокислот в почках и кишечнике.
4. Вторичные гипераминоцидурии: синдром Фанкони, фруктоземия, галактоземия, болезнь Вильсона-Коновалова и др. При данных состояниях возникает вторичная генерализованная гипераминоацидурия в результате вторичных тубулярных нарушений.
Впервые описана в 1934 г. Folling под названием "фенилпировиноградная имбецильность". Тип наследования - аутосомнорецессивный. Частота заболевания составляет 1:10000- 1:20000 новорожденных. Пренатальный диагноз возможен при использовании генетических зондов и биопсии ворсин хориона.
К развитию классической клинической картины при ФКУ приводит недостаточность фенилаланингидроксилазы и недостаточность редуктазы дигидроптерина- 2-го фермента, обеспечивающего гидроксилирование фенилаланина. Их недостаток приводит к накоплению фенилаланина (ФА) в жидких средах организма (схема 1). Как известно, ФА относится к незаменимым аминокислотам. Поступающий с продуктами питания и не используемый для синтеза белка, он распадается по тирозиновому пути. При ФКУ наблюдается ограничение превращения ФА в тирозин и, соответственно, ускорение его превращения в фенилпировиноградную кислоту и другие кетоновые кислоты.
Схема 1. Варианты нарушений метаболизма фенилаланина.
Существование различных клинико-биохимических вариантов ФКУ объясняется тем, что фенилаланингидроксилаза является частью мультиферментной системы.
Различают следующие формы ФКУ:
1. Классическая
2. Скрытая.
3. Атипичная.
Развитие атипичных и скрытых форм ФКУ связывают с недостаточностью фенилаланинтрансаминазы, тирозинтрансаминазы и оксидазы парагидроксифенилпировиноградной кислоты. Атипичная ФКУ обычно не сопровождается поражением нервной системы в результате позднего развития ферментативного дефекта.
У женщин с фенилкетонурией возможно рождение детей с микроцефалией, задержкой умственного развития, нарушениями развития мочевыделительной системы, поэтому необходимо назначение диетотерапии во время беременности.
Клинические симптомы у больных ФКУ
При рождении ребенок с фенилкетонурией выглядит здоровым. Заболевание у этих детей проявляется на первом году жизни.
1. Интеллектуальный дефект. Нелеченный ребенок теряет около 50 баллов IQ к концу 1-го года жизни. У больных не выявляется зависимости между уровнем ФА и степенью интеллектуального дефекта.
2. Судорожный синдром (4 50%), экзема, гипопигментация.
3. Нарушение координации движения.
4. Задержка развития статических и двигательных функций.
5. Поражение пирамидных путей и стриопаллидарной системы. Клинические проявления классической ФКУ редко встречаются в странах, в которых действует программа неонатального скрининга на это заболевание.
У детей с фенилкетонурией наблюдается повышенный уровень в моче метаболитов ФА. Увеличение в физиологических жидкостях содержания ФА и недоокисленных продуктов его метаболизма приводит к поражению нервной системы. Определенная роль в этих нарушениях принадлежит дисбалансу аминокислот (дефицит тирозина, который в норме активно участвует в построении белкового компонента миелина). Демиелинизация является характерным патоморфологическим признаком фенилкетонурии. Нарушение соотношения аминокислот в
крови приводит к нарушению уровня свободных аминокислот в головном мозге, что вызывает слабоумие, гиперкинезы и другие неврологические симптомы.
Пирамидные симптомы обусловлены нарушением процессов миелинизации. Избирательный характер поражения нервной системы объясняется особенностями миелинизации- поражаются наиболее молодые в филогенетическом отношении отделы, выполняющие сложные и дифференцированные функции. С недостаточным образованием меланина из тирозина связывают голубой цвет глаз, светлую кожу. Запах "плесени" ("мышиный", "волчий") объясняется наличием фенилуксусной кислоты в моче. Кожные проявления (экссудативный диатез, экземы) связаны с выделением аномальных метаболитов. Недостаточность образования адренергических гормонов из тирозина приводит к артериальной гипотонии.
Необходимо отметить, что при ФКУ в патологический процесс вовлекается печень, но характер морфологических расстройств не является специфичным: выявляются признаки тканевой гипоксии, нарушения окислительной и белоксинтезирующей функции, перегрузка липидами. Наряду с этим наблюдаются компенсаторно-приспособительные изменения: высокое содержание гликогена, гиперплазия митохондрий. Генерализованную гипераминоацидемию при ФКУ можно объяснить вторичным нарушением метаболизма аминокислот в связи с повреждением гепатоцитов, т.к. многие ферменты, участвующие в аминокислотном обмене, локализуются в печени.
У нелеченых больных с классической ФКУ наблюдается значительное снижение концентрации катехоламинов, серотонина и их производных в моче, крови, ликворе. Поэтому в комплексном лечении ФКУ необходима промедиаторная коррекция, так как парциальный интеллектуальный дефект может быть связан с нейромедиаторными нарушениями.
Критерии диагностики классической формы фенилкетонурии:
1. Уровень ФА в плазме выше 240 ммоль/л.
2. Вторичный дефицит тирозина.
3. Повышенный уровень в моче метаболитов ФА.
4. Сниженная толерантность к полученному внутрь ФА.
Методы диагностики фенилкетонурии:
1. Проба Феллинга с FeCl 3 - при положительном анализе появляется сине-зеленое окрашивание мочи.
2. В крови выявление избытка фенилаланина возможно с помощью бактериального экспресс- теста Гольдфарба или теста Гатри (т.к. в течение первых дней жизни фенилпировиноградная кислота в моче может отсутствовать).
При ФКУ проводится лечение диетой с ограниченным содержанием ФА (главным образом назначают овощные блюда, мед, фрукты). Такие продукты, как молоко, молочные изделия, яйца, рыба, должны быть полностью исключены в период пребывания больных с ФКУ на острой диете. Назначаются специальные препараты (цимогран, лофеналак) и витамины.
Оптимальные сроки обследования новорожденных- 6-14 день жизни, начало терапии - не позднее 21 дня жизни. Необходимо помнить, что проведение исследования в первые сутки не исключает ложноположительных или ложноотрицательных результатов (повторное исследование проводят до 21 дня жизни). Эффективность лечения оценивается по интеллектуальному уровню развития пациента. Необходимо отметить, что лечение, начатое после года не нормализует интеллект полностью (возможно, это связано с развитием необратимых изменений в мозге).